Prediction of potential miRNA–disease associations based on stacked autoencoder


![]()
Abstract
近年来,越来越多的生物学实验和科学研究表明,小 RNA (miRNA)在人类复杂疾病的发展中起着重要作用。因此,发现 miRNA 与疾病的关联有助于准确诊断和有效治疗疾病。通过基于生物学数据的计算方法识别 miRNA 与疾病的关联已被证明是低成本和高效率的。在这项研究中,我们提出了一个计算模型称为堆叠自动编码器用于潜在的 miRNA-疾病关联预测(SAEMDA)。在 SAEMDA,所有的 miRNA 疾病样本都被用于无监督地预训练一个堆叠的自编码器(SAE)。然后,在 SAE 中增加一个带有 softmax 分类器的输出层,利用正样本和相同数量的选定负样本对 SAE 进行监督微调(fine-tune)。SAEMDA 可以充分利用所有未标记的 miRNA 疾病对的特征信息。因此,SAEMDA 适用于包含小标记样本和大未标记样本的数据集。结果表明,SAEMDA 在全局和局部留一交叉验证中的 AUC 分别为0.9210和0.8343。此外,SAEMDA 经过100次5折交叉验证,平均得到0.9102 ±/-0.0029的 AUC 和标准差。这些结果比以前的模型要好。此外,我们进行了三个案例研究,以进一步证明 SAEMDA 的预测准确性。因此,前50位预测的 miRNA 中有82% (乳腺肿瘤)、100% (肺肿瘤)和90% (食管肿瘤)得到了数据库的验证。因此,SAEMDA 可能是一个有用和可靠的模型来预测潜在的 miRNA 疾病的关联。
Keywords: microRNA, disease, association prediction, Stacked Autoencoder, pretraining, fine-tuning
目录
1.引言
虽然上述模型在一定程度上表现出了可靠的性能,但每种模型都有其自身的局限性,需要进一步改进。由于深度学习技术可以更好地学习数据的表示,并且近年来已经成功地用于基因组学和药物发现等许多领域[40] ,我们考虑将其应用于 miRNA 疾病关联的预测。此外,只有具有已知标签的对才能用于训练普通的多层感知机网络,因此我们需要通过使用所有 miRNA-disease 对来预训练多层感知机网络,以在一定程度上减少已知关联过少对预测准确性的影响。受 Bahi 等人[41]的启发,我们提出了一个用于潜在的 MiRNA-疾病关联预测的堆叠自动编码器模型(SAEMDA),该模型利用了深度学习和预训练。我们首先使用所有 miRNA 疾病对以无监督的方式预训练堆叠自动编码器(SAE)。然后,利用正样本和相同数量的随机选择的负样本,以监督的方式对 SAE 进行微调。通过三种交叉验证方法对该方法的预测性能进行了评价。结果,SAEMDA 在全局留一交叉验证(LOOCV)中获得了0.9210的 AUC,在局部 LOOCV 中获得了0.8343的 AUC,以及在100倍5倍交叉验证中的平均 AUC 和标准差为0.9102 ± 0.0029。此外,我们还进行了三个案例研究来验证 SAEMDA 的预测精度。在乳腺肿瘤(BN) ,肺肿瘤(LN)和食管肿瘤(EN)的三种不同类型的病例研究中,前50名预测的潜在相关 miRNA 中的41,50和45通过数据库验证。
2.结果
3.讨论
预测潜在的 miRNA-疾病关联使研究人员能够更好地了解疾病的机制,促进复杂疾病的诊断、治疗和预后。在这项研究中,我们开发的 SAEMDA,可以有效地补充传统的生物实验方法。在 SAEMDA,所有的 miRNA 疾病样本都被用于预训练一个SAE。然后,用阳性样品和相同数量的阴性样品对 SAE 进行微调。SAEMDA 在三种类型的交叉验证中均优于其他模型。SAEMDA 方法优于以往的方法,主要是因为它在训练过程中充分利用了所有未标记样本的信息。此外,三种实例分析的结果进一步说明了 SAEMDA 的可靠性预测性能。除了 miRNA- 疾病关联预测之外,生物信息学领域还有许多重要的关联预测问题,如 lncRNA- 疾病关联预测[58] ,环状 RNA (circRNA)-疾病关联预测[59]和蛋白质-蛋白质相互作用预测[60]。在 miRNA-疾病关联预测任务中,SAEMDA 表现出良好的性能。因此,可以考虑利用 SAEMDA 框架来解决上述链路预测问题。
SAEMDA 的可靠性归功于以下几个方面。首先,我们研究中使用的数据包含495个 miRNA 和383个疾病的189585个 miRNA 疾病对,只有5430个已知的关联。由于 SAEMDA 采用了无监督预训练和监督微调相结合的方法,因此特别适合于由大量未标记数据和少量标记数据组成的数据集。预训练过程使模型能够了解所有 miRNA-疾病对的特征,并弥补了传统的监督式学习模型只能通过标签样本进行训练的缺陷。此外,微调过程使模型能够学习少量标记数据的标签信息,以进一步提高性能。其次,SAEMDA 整合了不同的相似性网络,使特征能够更好地捕获所有 miRNA-疾病对的信息。最后,我们在 SAEMDA 的训练过程中选择了 Adam 优化器,因为它比传统的随机梯度下降(SGD)优化器更有效率。
然而,SAEMDA 仍有一些局限性。首先,神经网络的超参数(如隐层数和每层神经元数)没有很好地确定。其次,SAEMDA 在100次5折交叉验证中获得了比对照模型大的标准差。因此,SAEMDA 模型在稳定性方面略逊于其他模型,这是深度学习中的一个常见问题。第三,微调过程中需要正、负样本,但随机选择未标记样本作为负样本会带来不准确的信息。最后,在作为疾病-miRNA 对特征的疾病和 miRNA 的剪接相似性方面还有改进的余地。因此,如何构建和提取 miRNA-疾病对的可靠特征将是未来预测方法设计的研究方向。此外,有必要设计适当的方法来改变阴性样本的选择方式。聚类算法可以考虑用于负样本选择过程[61-63]。此外,设计有效的方法引入其他生物学信息来帮助预测潜在的 miRNA-疾病关联也是一个重要的方向。
4.材料和方法
4.1.材料
首先,从 HMDD v2.0数据库获得人类 miRNA-疾病关联[42]。具体来说,有495种 miRNA,383种疾病和5430种实验证实的 miRNA 与疾病的关联。我们使用 nd 和 nm 分别表示疾病和 miRNA 的数量。大小为 nm × nd 的邻接矩阵 A 被用来代表所有的 miRNA-疾病对。如果 miRNAm (i)与疾病 d (j)相关,则 A (i,j)等于1; 否则为0。此外,miRNA 的功能相似性评分是在之前的研究中计算出来的[64] ,可以从 http://www.cuilab.cn/files/images/cuilab/misim.zip 下载。用 FS 矩阵表示 miRNA 功能相似矩阵。此外,我们通过有向无环图描述了两种疾病之间的关系,并根据以前的研究使用了两种不同的方法来计算疾病的语义相似度[32]。基于两种疾病的 DAGs 的共同部分越大,语义相似度值越大的假设,我们通过以前的研究[32]中的方法计算了第一类疾病语义相似度矩阵 SS1。由于同一层 DAG 中的不同疾病术语对所研究疾病的语义价值应该有不同的贡献,我们重新定义了每个疾病术语的语义贡献,并进一步计算了第二类疾病语义相似性矩阵 SS2[32]。此外,基于相似疾病(miRNA)与 miRNA (疾病)具有相似的相互作用和非相互作用模式的假设,我们根据以前的方法计算了疾病(miRNA)的高斯相互作用谱核相似矩阵 KD (KM)[65]。值得注意的是,在每轮 LOOCV 和5折交叉验证中,KD 和 KM 将根据除测试样本以外的所有已知关联信息重新计算。最后,我们将 miRNA 的高斯互作谱核相似性与 miRNA 功能相似性结合起来,根据以前的研究[22]的方法得到完整的 miRNA 相似性矩阵 SM 如下:
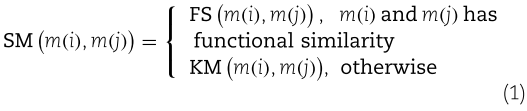
同样,我们也通过整合高斯相互作用轮廓核相似度和两种疾病语义相似度计算了综合疾病相似度矩阵 SD。

4.2.SAEMDA

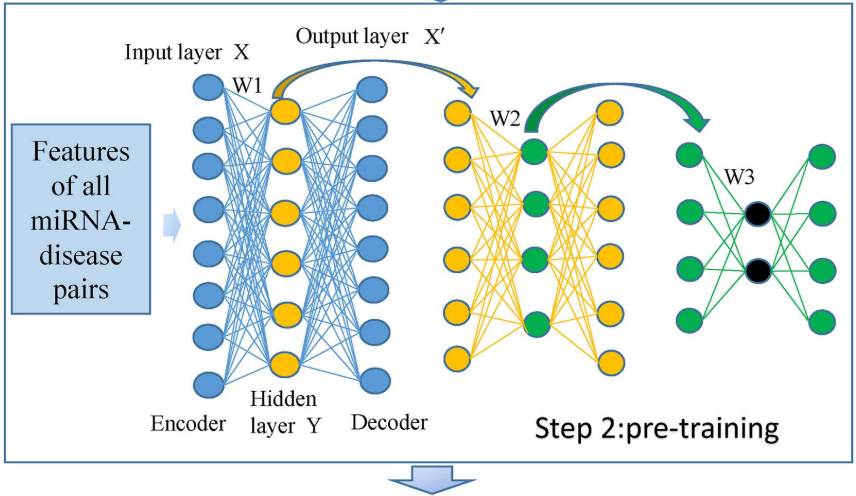
在这项研究中,我们提出了一个新的模型命名为 SAEMDA 来预测潜在的 miRNA 疾病的关联。SAEMDA 的流程图如图2所示。SAEMDA 的第一步是数据准备,即将 miRNA-disease 对表示为特征向量。如前文所述,我们构建了 miRNA-疾病对(nm × nd)的邻接矩阵 A,整合的 miRNA 相似性矩阵 SM (nm × nm)和整合的疾病相似性矩阵 SD (nd × nd)。从中分别提取各种 miRNA 和疾病的 nm 和 nd features。连接所研究的疾病和miRNA的特征向量产生了每个 miRNA-疾病对的 nm + nd features。在所有的 miRNA 疾病对中,共有5430对是已知的关联,其余的 miRNA 疾病对是未标记的。
SAEMDA 的第二步是基于所有 miRNA 疾病对的 SAE 的无监督预训练。SAE 的深度学习模型可以通过叠加多个自动编码器(AE)[66]来构建。AE由编码器和解码器组成。编码器通过将输入特征从输入层映射到隐藏层来学习新的表示,而解码器则从隐藏层到输出层重构原始输入。此外,输入层和输出层具有相同数量的神经元。AE可以降低原始数据的维数。将训练样本的特征向量 X 输入 AE 后,定义隐层的表示形式如下:
![]()
其中 σ、 W 和 b 分别代表编码器的激活函数(我们研究中的 tanh)、权重矩阵和偏置向量。然后,根据对隐藏层的表示,重新构造了与 X型状相同的输出 ![]() :
:
![]()
其中 ![]() 和
和 ![]() 表示解码器的权重矩阵和偏置向量。接下来,基于 Adam 优化器对 AE 进行了最小化重建损失的训练:
表示解码器的权重矩阵和偏置向量。接下来,基于 Adam 优化器对 AE 进行了最小化重建损失的训练:
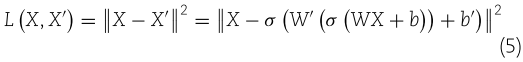
在这项研究中,根据以前的研究[41] ,SAE 是通过叠加三个 AE 构建的。对 SAE 进行了以下无监督的预训练:
1)利用所有 miRNA 疾病对的特征向量对一个 AE 进行训练。
2)去除AE中的解码层。然后,以第一个 AE 生成的特征向量作为输入,构造一个新的AE。
3)新的AE被训练,而以前训练的 AE 的权重和偏差保持不变。
4)重复步骤2和步骤3,直到三个 AEs 堆积起来。
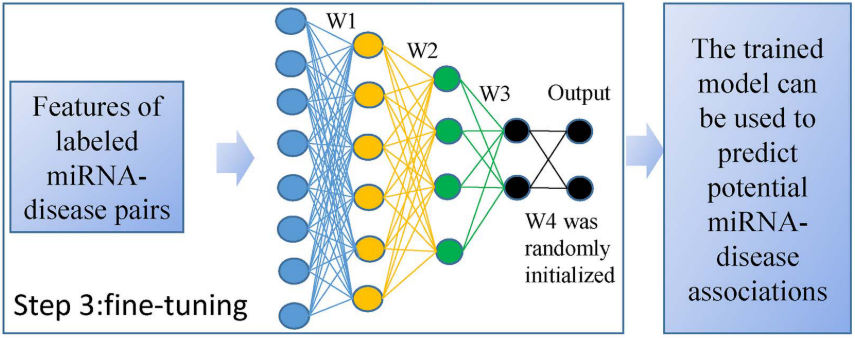
经过无监督预训练,我们得到了 SAE 的权重矩阵 W1、 W2和 W3,以及 b1、 b2和 b3的偏置向量。然后,SAEMDA 的第三步是基于正负样本的 SAE 监督微调。在这里,5430个已知的 miRNA 疾病关联被作为阳性样本。另外,从未标记的 miRNA 疾病对中随机选择了5430个阴性样本。微调过程包括以下步骤:
1)在预训练过程中得到的 SAE 中增加了输出层。这里,输出层和前一层之间的权重矩阵 W4和偏置向量 b4被随机初始化。
2)采用阳性样本和相同数量的阴性样本进行训练。
最后,训练后的 SAE 可用于预测潜在的 miRNA-疾病关联。值得注意的是,SAEMDA 在每个隐藏层都使用了 tanh 激活函数,在输出层使用了 softmax 分类器。此外,在微调过程中采用交叉熵作为损失函数,并利用Adam优化器对 SAE 进行优化。此外,我们将三个 AE 的隐藏层数分别设置为512、256和128。在设置模型的超参数后,我们用0.0001的学习率训练 SAEMDA 以获得最终的 miRNA-疾病关联评分。