DAEMKL:Predicting miRNA–Disease Associations Through Deep Autoencoder With Multiple Kernel Learning(发表于IEEE Transactions on Neural Networks and Learning Systems)

![]()
目录
2.材料和方法(MATERIALS AND METHODS)
D.多核学习(Multiple Kernels Learning)
E.特征表示的回归模型(Regression Model for Feature Representation)
3.结果与讨论(RESULTS AND DISCUSSION)
A. 实现细节(Implementation Details)
B.性能评价(Performance Evaluation)
C.多核学习的效果(Effect of Multiple Kernels Learning)
摘要(Abstract)
确定microRNA(miRNA)-疾病关联是预防、诊断和治疗复杂疾病的重要组成部分。 然而,湿实验识别MDAs效率低且昂贵。 因此,建立可靠有效的数据集成模型预测MDAs具有重要意义。 本文提出了一种基于多核学习的深度自动编码器(DAEMKL)的MDAs预测方法。 首先,DAEMKL在miRNA空间和疾病空间分别应用多核学习(MKL)构建miRNA相似度网络和疾病相似度网络。 然后,通过回归模型从miRNA相似度网络和疾病相似度网络中学习每个疾病或miRNA的特征表示。 然后将整合后的miRNA特征表示和疾病特征表示输入到深度自动编码器(DAE)中。 此外,通过重构误差预测新的MDAs。 最终,AUC的结果表明,DAEMKL实现了出色的性能。 此外,三种复杂疾病的案例研究进一步证明,DAEMKL具有卓越的预测性能,能够发现大量潜在的MDAs。 总的来说,我们的方法DAEMKL是一种有效的MDA识别方法。
索引术语-深度自动编码器(DAE),特征表示,microRNA(miRNA)-疾病关联(MDAs),多核学习(MKL)。
1.引言(Introduction)
mIcroRNAs(miRNAs)是一组长度约为22个核苷酸的非蛋白编码核糖核酸(RNAs),是多种生物过程的关键调节因子[1]-[3]。 miRNA及其靶mRNA的生物合成和功能障碍可能导致各种疾病[4]。 研究表明,miRNAs与乳腺癌、胰腺癌、淋巴瘤等多种恶性疾病的预防、诊断和治疗密切相关[5]、[6]。 因此,研究miRNA与疾病的关联在生物医学中具有重要意义。 然而,传统的生物实验方法鉴定MDAs费时费力,需要设备。 因此,越来越多的研究者转向生物信息学的方法来分析和预测新的MDAs。
以往预测MDAs的计算方法主要归纳为三类[7]。 第一类是基于相似性度量的方法,基于相似疾病更有可能与相似的miRNAs相关,反之亦然的假设。 Xuan等人[8]提出了预测MDAs的加权k最相似邻居。 该方法综合疾病术语和疾病表型相似性,可有效预测MDAs。 陈等人[9]提出了一种基于随机游走重启的全局网络相似性度量方法,用于预测新的MDAs(RWRMDA)。 不久之后,施等人[10]提出了另一种新的随机游走方法来预测MDAs。 该方法将疾病基因和miRNA靶基因映射到蛋白质相互作用网络中,利用随机游走法预测MDAs。 You等人[11]提出了一种基于路径的预测MDAs(PBMDAs)的方法。 PBMDA构造了一个异构网络,然后采用深度优先搜索方法预测新的MDAs。 由于考虑了网络拓扑结构,PBMDA取得了很好的预测效果。 Xiao等人[12]提出了一种利用自适应多源多视图潜在特征识别新MDAs(M2LFL)的新方法。 M2LFL采用自适应联合图正则化项将MiRNAs的与疾病的manifold structures结合起来。 最后,该方法取得了较好的预测效果。
第二类是基于机器学习的方法。 近年来,机器学习在各个领域都很受欢迎。 多种预测MDAs的机器学习模型也得到了众多学者的认可。 2013年,Jiang等人[13]提出了一种基于支持向量机(SVM)的方法。 他们从每个MDA数据中提取特征,用于训练SVM分类器,以挖掘未知MDAs的潜在信息。 Chen等人 [14]实现了集成学习和链接预测来发现MDAs(ELLPMDAs)。ELLPMDA通过集成学习综合了三种算法得到的预测结果。 此外,Chen等人[15]提出了另一种方法,即用归纳矩阵补全法预测潜在的MDAs(IMCMDAs)。IMCMDA的动机是使用已知的MDAs信息、miRNA相似性和疾病相似性信息来补充缺失的关联。还采用了一些矩阵分解方法来预测MDAs。 Xiao等人提出了一种基于图正则化非负矩阵分解(GRNMF)的MDAs预测框架。 最近,Gao等人[16]发展了另一种发现新MDAs的矩阵分解方法,称为基于最近谱的协同矩阵分解(NPCMF)。 NPCMF考虑了相似度网络的邻居信息,对miRNA相似度矩阵和疾病相似度矩阵施加了最近的谱,取得了良好的预测效果。
第三类是基于深度学习的方法。 随着深度学习方法在自动驾驶、人脸识别等领域的成功应用[17]-[19],研究人员开始开发一些关于MDA预测的深度学习方法。 例如,chen等人[20]提出了一种基于深度信念网络的MDAs预测方法(DBNMDAs)。 DBNMDA首先从所有miRNA-疾病对中提取特征来训练受限玻尔兹曼机器,然后选择与正样本数量相同的负样本来调整深度信念网络。 此外,Ji等人[21]提出了一种基于自动编码器的预测新MDAs(AEMDAs)的计算方法。 AEMDA通过深度学习算法训练了三种模型,包括miRNA模型、疾病模型、深度自动编码器(DAE)模型。 AEMDA首先训练miRNA模型和疾病模型获得特征表示,然后输入AutoEncoder完成MDAS的预测。 最后,AEMDA取得了较好的预测性能。
尽管上述方法取得了优异的性能,但它们仍然面临着一些局限性。 一方面,现有数据库中只有少量已知关联作为阳性样本,其余的样本都是未知关联,因此没有经过验证的阴性样本。 因此,一些有监督的机器学习算法很难训练出可靠的预测模型。 另一方面,在构建miRNA相似度核和疾病相似度核的过程中,大量方法只是简单地将高斯核与miRNA功能相似度核和疾病语义相似度核结合起来[16]。
为了克服上述不足,本研究通过多核学习的深度自动编码器(DAEMKL)开发了一个更完整的预测MDAs的深度学习框架。 在本研究中,DAE用于学习潜在的疾病相关miRNAs的特征,而不需要阴性样本。 最重要的是,DAEMKL集成了来自miRNA特征空间和疾病特征空间的多种信息。 几个核分别由miRNA和疾病空间构建。 然后将多核学习(MKL)应用于miRNA空间和疾病空间,构造miRNA相似度网络和疾病相似度网络。 基于多个不同信息源的MKL可以包含更多的先验信息,有利于捕捉深度交互。 对于每种疾病或 miRNA,其特征表示是通过回归模型从 miRNA 相似性网络和疾病相似性网络中学习得到的。MIRNA和疾病特征表示以类似的方式学习,以确保后续学习过程中数据的兼容性。 之后,将整合后的miRNA特征表示和疾病特征表示输入DAE。 最后,DAEMKL对输入数据进行重构,根据重构误差预测新的MDA。 因此,采用五折交叉验证(5-CV)和全局留一交叉验证(LOOCV)来验证预测性能。 此外,还进行了三种疾病的病例研究,以验证预测的新MDAS是否已被记录。 DAEMKL充分利用了先验知识和特征信息的兼容性,取得了良好的预测性能,挖掘出了更多未知的MDA。 本文的主要贡献概括如下。
1)开发了一个完整的基于深度学习的端到端预测框架,能够有效识别新的MDAs。
2)DAE作为预测器,可以有效地从已知的MDAs中学习特征信息,而不需要负样本。
3)将MKL引入到预测框架中,学习多源信息,使miRNAs和疾病的信息更加丰富。
本文的组织方式如下:第二节展示了数据集和用于预测MDAs的DAEMKL模型。 第三节介绍了实验结果和评价方法。 此外,我们的模型也与其他先进的方法进行了比较。 第四节对全文进行了总结。
2.材料和方法(MATERIALS AND METHODS)
A.已证实的人MDAs(已证实的人MDA )
已知的MDAs是通过人类小RNA疾病数据库(HMDD)V2.0[22]和HMDD V3.2[23]获得的。 HMDD是一个由研究人员手工收集的MDA数据库。 每个记录的MDA都包含详细的信息,不仅包括miRNA和疾病名称,还包括相关说明和参考文献的PubMed ID。 第一个数据集(D1)被认为是预测MDAs的金标准数据集,包括495个miRNA,383个疾病和5430个已证实的MDA。 第二个数据集(D2)也广泛应用于MDA预测[24],[25],包含550个miRNA,328个疾病和6088个MDA。 使用HMDD V3.2验证模型预测的新MDA是否被记录。 考虑到模型的适用性,DAEMKL模型和其他先进的方法使用D1和D2进行评估,以验证DAEMKL模型的性能。
B. MiRNA Space
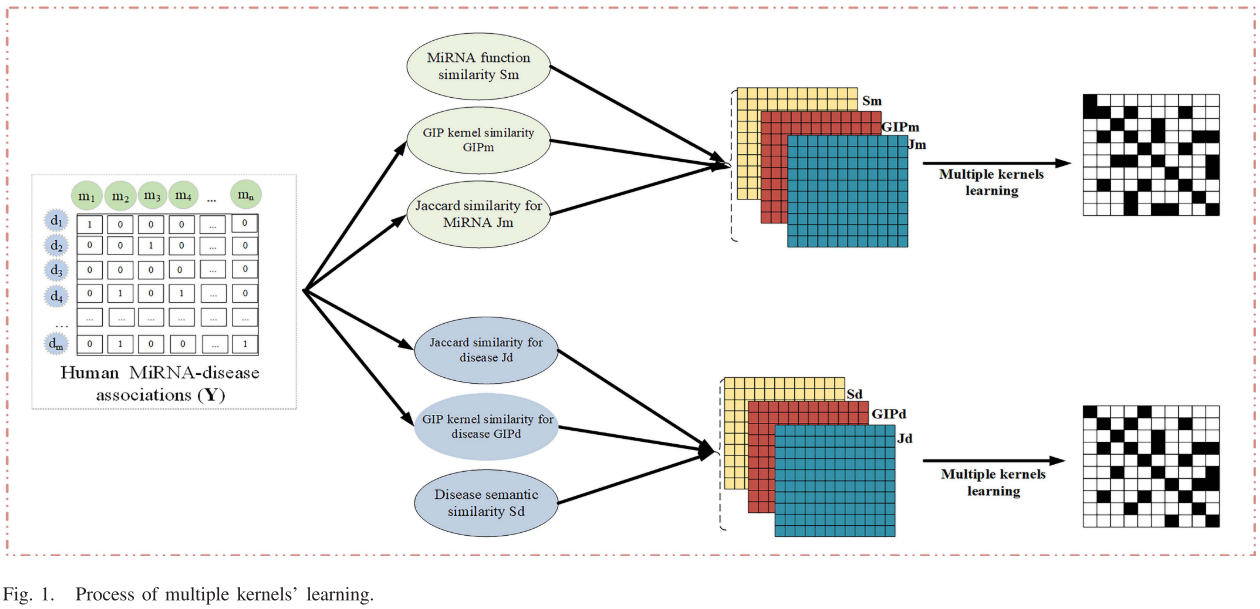
为了表征不同信息源的miRNAs的特征,构造了miRNAs的三种相似核,包括miRNAs功能相似度、高斯互作谱(GIP)核相似度和Jaccard相似度。
miRNA功能相似性从MISIM数据库中获得[26]。 MISIM数据库(http://www.cuilab.cn/files/images/cuilab/misim.zip)由Wang等人开发。 基于功能相似的miRNAs更可能与表型相似的疾病相关。 由此构造了miRNA功能相似矩阵![]() ,其中
,其中![]() 表示miRNA i 和miRNA j 的功能相似度。
表示miRNA i 和miRNA j 的功能相似度。
为了获得miRNA空间的网络相似度,根据前人的研究[27]计算了miRNA的GIP核相似度![]() 。 miRNA i 和miRNA j 之间的GIP核相似度定义为
。 miRNA i 和miRNA j 之间的GIP核相似度定义为
![]()
其中![]() 是miRNA i 和miRNA j 的关联谱,即分别是关联矩阵的第i列和第j列。 此外,θm是内核带宽的可调参数,定义如下:
是miRNA i 和miRNA j 的关联谱,即分别是关联矩阵的第i列和第j列。 此外,θm是内核带宽的可调参数,定义如下:
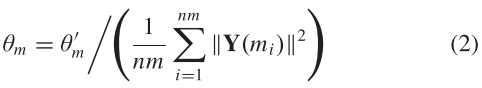
其中nm是miRNAs的数目,θm通常取为1[27]。
Jaccard相似度在文本挖掘和数据聚类等任务中得到了广泛的应用。 在这里,我们采用它来度量miRNAs之间的相似性。 公式如下:

C. Disease Space
在疾病空间中,相似核也包括三类:疾病语义相似度、GIP核相似度和Jaccard相似度。
语义相似度被认为是衡量疾病之间关系的可靠指标[28]。 根据前人的研究[26],基于医学主题词(Mesh)数据库开发了有向无环图(DAG)来计算疾病的语义相似度![]() 。DAG中有大量的点和有向边,其中每个点是一个疾病,每个有向边表示疾病之间的关系。
。DAG中有大量的点和有向边,其中每个点是一个疾病,每个有向边表示疾病之间的关系。
根据相同的计算规则,GIP内核相似度![]() 和Jaccard相似度
和Jaccard相似度![]() 如下所示:
如下所示:
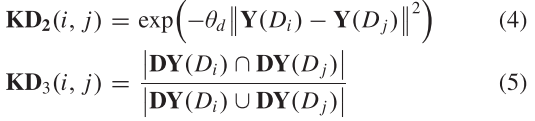
其中![]() 是疾病I和疾病J的关联谱,分别表示关联矩阵的第i行和第j行。
是疾病I和疾病J的关联谱,分别表示关联矩阵的第i行和第j行。 ![]() 表示关联图谱中与疾病 j 相关的miRNAs集合。
表示关联图谱中与疾病 j 相关的miRNAs集合。
D.多核学习(Multiple Kernels Learning)
考虑到单个相似核或miRNA功能相似矩阵与GIP核相似矩阵的简单组合不能表示miRNA之间的相似信息,采用多核学习(MKL)来获得最优核[29]。 以图为例说明了MKL的过程 1. MKL通过一个优化算法计算每个核的权重,从而得到一个miRNA的集成核。
miRNAs的最优核![]() 定义为
定义为
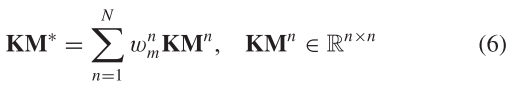
其中![]() 是miRNA核的最优权值,N是miRNA核的个数。 MKL的学习过程是利用理想核对集成核进行优化。 公式如下:
是miRNA核的最优权值,N是miRNA核的个数。 MKL的学习过程是利用理想核对集成核进行优化。 公式如下:

其中,![]() 表示Frobenius范数,
表示Frobenius范数,![]() ,β表示正则化参数。 上述优化问题可计算如下:
,β表示正则化参数。 上述优化问题可计算如下:

同样,通过上述MKL算法也可以计算出疾病核的权重![]() 。 疾病的最终最优核
。 疾病的最终最优核![]() 可得到如下:
可得到如下:
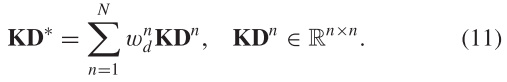
E.特征表示的回归模型(Regression Model for Feature Representation)
受 Ji 等人的工作启发。 [21]中,通过回归模型从最终的最优相似度核学习miRNA特征表示和疾病特征表示。 在miRNA空间中,对于每一个miRNA,我们构造了一个高维向量![]() 来表示高维空间中的特征。 所有高维向量
来表示高维空间中的特征。 所有高维向量![]() 的集合构成miRNA的特征矩阵
的集合构成miRNA的特征矩阵![]() 。 特征矩阵
。 特征矩阵![]() 可以表示如下:
可以表示如下:
![]()
其中![]() 表示i th miRNA的向量,kM 表示
表示i th miRNA的向量,kM 表示![]() 的大小,nm表示miRNA的数目。 在回归模型的训练过程中,特征矩阵
的大小,nm表示miRNA的数目。 在回归模型的训练过程中,特征矩阵![]() 随机初始化。
随机初始化。
此外,余弦相似度被用作测量![]() 之间距离的一种可靠有效的方法。 回归模型采用最优相似核
之间距离的一种可靠有效的方法。 回归模型采用最优相似核![]() 作为 ground-truth 标签。 但是,余弦相似度计算值的取值范围为[-1,1],在
作为 ground-truth 标签。 但是,余弦相似度计算值的取值范围为[-1,1],在![]() 中的相似度值是非负的。 因此,余弦相似度得分的范围在0和1之间调整如下:
中的相似度值是非负的。 因此,余弦相似度得分的范围在0和1之间调整如下:
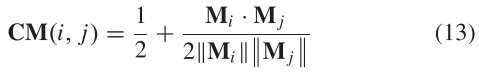
其中![]() 分别是miRNA i 和miRNA j 的特征表示向量,而
分别是miRNA i 和miRNA j 的特征表示向量,而![]() 表示这两个miRNA的距离度量。 然后,构造miRNA回归模型来学习miRNAs的特征表示,该模型使
表示这两个miRNA的距离度量。 然后,构造miRNA回归模型来学习miRNAs的特征表示,该模型使![]() 和最优相似核
和最优相似核![]() 最小化。 公式如下所示:
最小化。 公式如下所示:

其中![]() 是训练样本的数目。 在训练过程中,采用平方损失作为反向传播的标准,并采用随机梯度下降(SGD)方法更新特征矩阵
是训练样本的数目。 在训练过程中,采用平方损失作为反向传播的标准,并采用随机梯度下降(SGD)方法更新特征矩阵![]() 。 经过多次训练,得到了可靠的miRNA特征矩阵
。 经过多次训练,得到了可靠的miRNA特征矩阵![]() 。
。
类似地,疾病![]() 的可靠特征表示可以通过回归训练。 公式定义如下:
的可靠特征表示可以通过回归训练。 公式定义如下:

其中 nd 为疾病数,KD 表示向量大小。 在训练过程中,我们将 kD=kM 设置为0.5。 CD是疾病的余弦相似度量,![]() 也是训练样本数。
也是训练样本数。
F.深度自动编码器
一个典型的自动编码器被定义为一个无监督学习模型,其目的是重建输入特征[30],[31]。 近年来,DAE是一种常用的深度学习方法,在特征提取、模式识别等领域得到了广泛的应用[32]-[34]。 因为数据集中只有一小部分关联是被证实的,我们开发了一种基于DAE的半监督学习方法。 我们的模型分为编码和解码两个部分。 在编码部分,将miRNA的高维特征向量![]() 和疾病的高维特征向量
和疾病的高维特征向量![]() 串联起来作为自动编码器的输入
串联起来作为自动编码器的输入![]() 。 然后编码后得到潜在变量
。 然后编码后得到潜在变量![]() 。 之后,DAE在解码部分根据潜在变量
。 之后,DAE在解码部分根据潜在变量![]() 输出重构
输出重构![]() 。
。
在训练过程中,已知的MDAs被认为是可观察的样本。 DAE的训练样本![]() 通过miRNA的特征向量
通过miRNA的特征向量![]() 和疾病的特征向量
和疾病的特征向量![]() 连接起来。 训练样本
连接起来。 训练样本![]() 定义如下:
定义如下:
![]()
其中![]() 是第i个训练样本。 对于样本
是第i个训练样本。 对于样本![]() ,编码过程的作用是将样本
,编码过程的作用是将样本![]() 的高维特征转换为潜在变量
的高维特征转换为潜在变量![]() 。 编码过程定义如下:
。 编码过程定义如下:
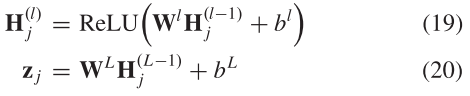
其中![]() 是编码器中隐藏层的数目,本文将
是编码器中隐藏层的数目,本文将![]() 设置为2。
设置为2。 ![]() 表示
表示![]() ,
,![]() 是第
是第![]() 隐层特征表示。
隐层特征表示。![]() 表示权重矩阵,
表示权重矩阵,![]() 表示偏差。 另外,非线性激活函数
表示偏差。 另外,非线性激活函数![]() 是获取表示一种可靠的方法。
是获取表示一种可靠的方法。
解码器的输出是根据潜在变量![]() 重构的特征表示
重构的特征表示![]() ,解码器定义如下:
,解码器定义如下:
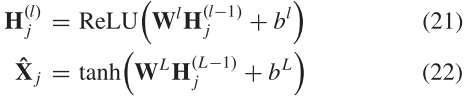
其中![]() 是编码器输出的潜变量
是编码器输出的潜变量![]() ,
,![]() 表示权重矩阵,
表示权重矩阵,![]() 表示第
表示第![]() 层中的偏置。
层中的偏置。 ![]() 是译码器对输入
是译码器对输入![]() 输出的重构,tanh(·)是非线性激活函数。
输出的重构,tanh(·)是非线性激活函数。
最后分两部分计算了损失函数。 第一部分是所有重建误差的平方损失之和,第二部分是一个正则化项。 因此,DAEMKL的损失函数可以表示为

其中![]() 是训练样本数,λ是超参数,
是训练样本数,λ是超参数,![]() 是Jacobian的正则化。 在训练的每个阶段,DAEMKL通过最小化
是Jacobian的正则化。 在训练的每个阶段,DAEMKL通过最小化![]() 的损失来更新模型中的所有参数。
的损失来更新模型中的所有参数。
G.DAEMKL
DAEMKL 的整个流程图如图2所示。我们的模型框架可以用四个步骤来描述。首先,采用 MKL 分别获得 miRNA 空间和疾病空间的最优核。其次,通过回归模型从miRNA空间和疾病空间的最优核学习每个miRNA和每个疾病的特征表示。 第三,将整合后的miRNA特征表示和疾病特征表示输入DAE。 最后,通过重构误差预测新的MDA。

3.结果与讨论(RESULTS AND DISCUSSION)
A. 实现细节(Implementation Details)
DAEMKL通过以下方案进行训练。 首先,以均方损失作为误差反向传播准则,采用SGD方法对回归模型进行端到端的训练。 此外,采用Adam算法对回归模型进行优化。 经过60个epoch,得到了M和D的两个特征表示。 此外,DAE是用M、D和已知的MDAs训练的,DAE通常在50-100个epoch后达到收敛。 我们采用M和D的串联向量作为输入,因此输入层有![]() 神经元。 编码器的隐层节点分别设置为
神经元。 编码器的隐层节点分别设置为![]() ,解码器的隐层节点分别设置为
,解码器的隐层节点分别设置为![]() 和
和![]() 。
。
此外,DAEMKL的初始学习速率![]() 、DAEMKL的训练epoch
、DAEMKL的训练epoch![]() 、向量大小
、向量大小![]() 和
和![]() 等超参数也影响DAEMKL的性能。 我们在数据库D1上使用这些参数的不同组合对DAEMKL进行训练,这些参数的范围为
等超参数也影响DAEMKL的性能。 我们在数据库D1上使用这些参数的不同组合对DAEMKL进行训练,这些参数的范围为![]() ∈[512,1024,2048,4096]、
∈[512,1024,2048,4096]、![]() ∈[50,100,150,200]和
∈[50,100,150,200]和![]() ∈[1,1e-1,1e-2,1e-3,1e-4]。 通过经验调整参数,我们在下面的实验中为DAEMKL设置
∈[1,1e-1,1e-2,1e-3,1e-4]。 通过经验调整参数,我们在下面的实验中为DAEMKL设置![]()
![]() 。
。
B.性能评价(Performance Evaluation)
在实验中,采用5-CV和全局LOOCV对DAEMKL的预测性能进行了评估。 对于5-CV,已知的MDAs被随机分为五个子部分。 每个子部分依次作为测试样本,其余四个子集作为训练集。 AUC的值是接收机工作特性(ROC)曲线下的面积。 AUC的值在0到1之间,其中0.5表示随机性能。 AUC值小于0.5表示预测结果无意义。 为了防止结果的偶然性,在每一轮中,都使用训练样本对DAEMDA进行训练。 然后我们计算剩余样本和未确认样本的重建误差。 跑50次结果的平均为最后的结果。
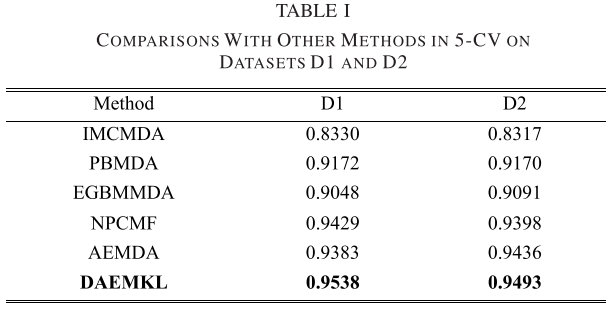
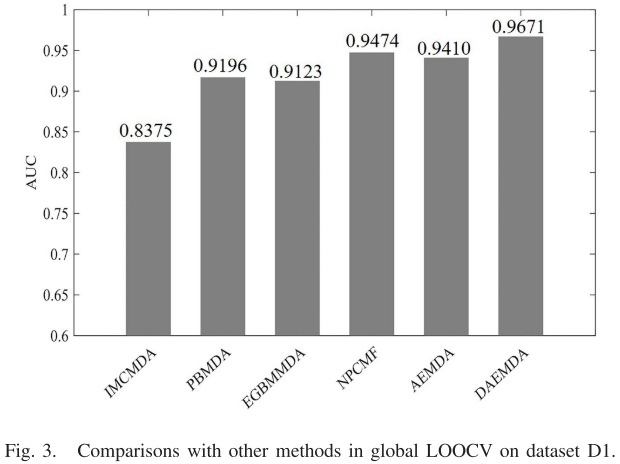
在应用5-CV和Global LOOCV对我们的方法进行评估时,我们还将DAEMKL与五种先进的MDA预测方法:IMCMDA[15]、PBMDA[11]、EGBMMDA[35]、NPCMF[16]和AEMDA[21]进行了比较。 为了将DAEMKL与最先进的方法进行比较,我们在两个数据集D1和D2上实现了5-CV,并在数据集D1上实现了全局LOOCV。 表一和图 3给出了DAEMKL的详细结果和比较方法。结果表明,DAEMKL 在 D1上获得了5-CV 为0.9583的 AUC 和全局 LOOCV 为0.9671的 AUC,两者与其他模型相比均排名第一。因此,DAEMKL 的性能明显优于其他五种最先进的方法。
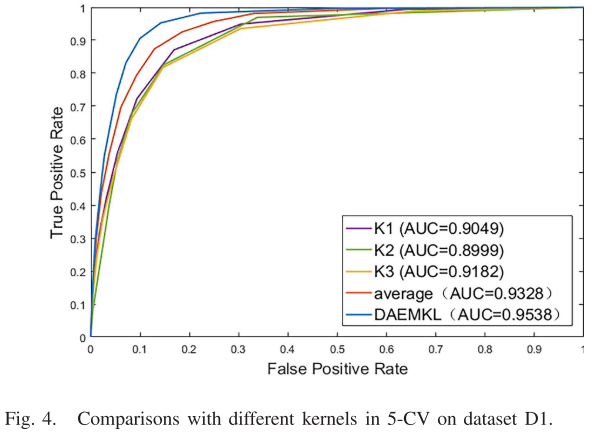
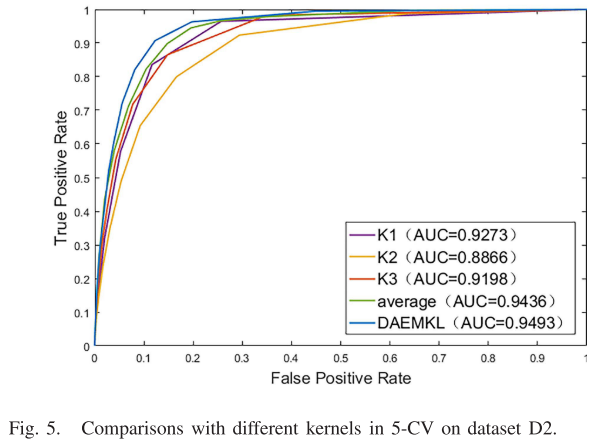
C.多核学习的效果(Effect of Multiple Kernels Learning)
在本研究中,多核学习被引入到我们的预测框架中,这有助于分别在miRNA空间和疾病空间提取高维特征。 通过比较DAEMKL和不使用MKL的模型(使用单个内核或平均内核)的性能,研究了MKL的性能。 平均核将GIP核与miRNA功能相似度核和疾病语义相似度核相结合。图4和5显示了不同核在5-CV中的性能比较。 实验标记K1为![]() ,或K2为
,或K2为![]() ,或K3为
,或K3为![]() 。
。
D. 案例研究(Case Studies)
为了进一步证明我们的方法的预测性能,并证实 DAEMKL 在现实情况下获得的预测结果的可靠性和合理性,我们对淋巴瘤,结肠新生物和胰腺肿瘤进行了病例研究。在实验中,D1数据集被用来训练我们的模型来考虑疾病的优先候选 miRNA。然后,选择与疾病相关的前30个潜在的miRNAs,并用MDA数据库的最新版本DBDEMC V2.0[36]和HMDD V3.2[37]对预测的miRNAs进行验证。
结肠肿瘤是胃肠道最常见的恶性肿瘤之一。 2018年中国肿瘤统计数据显示,我国结肠癌发病率和死亡率分别居所有恶性肿瘤的第三位和第五位。 为了评估DAEMKL的预测效果,我们选择结肠肿瘤作为我们的第一个病例研究。 如表II所列,96.67%的前30个miRNA与结肠肿瘤的关联已被这两个数据库验证。 在这30个新的关联中,60%的关联被DBDEMC V2.0和HMDD V3.2数据库确认。 许多研究证明,结肠肿瘤的发生发展与许多miRNAs密切相关。 例如,zHANG等人 [38]发现miR-21、miR-17和miR-19a促进结肠癌细胞的增殖和侵袭。

胰腺癌是一种高度致命的恶性肿瘤,存活率非常低。 大多数患者被诊断为晚期疾病,因此可用的治疗方法不太有效[40]。 因此,在胰腺肿瘤中识别潜在的miRNAs可以改善患者的预后。 本研究选取了前30个预测的胰腺癌相关miRNAs进行分析。 在筛选出的30个miRNA与胰腺癌的关联中,96.67%的关联可以在DBDEMC V2.0或HMDD V3.2数据库中找到证据。 另外,53.3%的胰腺癌与miRNA相关,只有miR-219未被证实。然而,Lahdaoui等人[39]发现 miR-219在胰腺癌细胞系中的过度表达导致细胞增殖和迁移减少。 具体详情载于表三。

淋巴瘤是一种起源于淋巴造血系统的恶性肿瘤,可累及身体的任何器官[40]。 2012年,全球约有386000名非霍奇金淋巴瘤患者和66000名霍奇金淋巴瘤患者,占癌症总数的3.2%[41]。 研究表明,miRNA生物标志物的存在对淋巴瘤的早期发现具有重要作用。 例如,张等人。 [42]在套细胞淋巴瘤中检测到miR-15a和miR-16下调。 此外,Li等人。 [43]发现miR-21水平的表达可作为弥漫性大B细胞淋巴瘤预后的一个新的生物标志物。 在本文中,我们将验证两个数据库是否记录了预测的MDA。 DAEMKL实现后,发现前30个新的miRNA与淋巴瘤的关联中有28个得到了DBDEMC V2.0和HMDD V3.2数据库的验证。 尽管两个数据库都证实了只有两种新的 miRNA,但我们在 HMDD v3.2中发现了相对较少的淋巴瘤记录。因此,挖掘更多与淋巴瘤相关的潜在 miRNA 对于淋巴瘤的预防和治疗具有重要意义。具体结果列于表四。

4. 结论(conclusion)
研究表明miRNAs与许多复杂疾病有关。 识别与疾病相关的潜在miRNAs对于了解疾病发病机制和疾病治疗具有重要意义。 本文提出了一种基于多核学习的DAE预测框架,用于预测新的MDA。 第一步是将MKL应用于miRNA空间和疾病空间,构造miRNA相似网络和疾病相似网络。 然后,对每个疾病或miRNA通过回归模型从miRNA相似度网络和疾病相似度网络中学习其特征表示。 最值得注意的是,集成的miRNA特征表示和疾病特征表示被输入到DAE中。此外,通过重构误差对新型 MDA 进行了预测。最后,将DAEMKL算法与5-CV中的五种最先进的方法进行了比较,结果表明,DAEMKL算法在预测MDAS方面具有良好的性能。 此外,还对三种复杂疾病进行了仿真实验,进一步验证了DAEMKL的性能。 因此,多种性能评价指标的结果表明,DAEMKL比现有的其他方法表现出了更好的预测性能。 它是识别潜在疾病相关miRNAs的可靠有效的深层模型。
总的来说,DAEMKL中也存在局限性。 首先,利用回归模型分别学习miRNA和疾病的特征表示。 因此,当数据库变得非常大时,模型的复杂性就不是很友好了。 此外,目前验证的MDAs的不足也可能影响我们的模型的预测能力。 在未来的工作中,我们将致力于开发更高效的特征表示方法。 此外,我们将尝试整合更多不同的数据库,以进一步提高我们的预测模型的性能。