Predicting miRNA-disease associations based on graph attention networks and dual Laplacian regularized least squares
源代码:GitHub - shine-lucky/MKGAT-main

![]()
Abstract
越来越多的生物医学证据表明,miRNA 的失调与人类的复杂疾病有关。疾病相关 miRNA 的鉴定对疾病的预防、诊断和治疗具有重要意义。为了减少生物医学实验的时间和成本,开发有效的计算方法来推断潜在的 miRNA 疾病关联有很强的动力。虽然已经提出了许多计算方法来解决这个问题,但是预测的准确性还需要进一步提高。在这项研究中,我们提出了一个计算框架 MKGAT 通过图注意网络(GATs)使用双拉普拉斯正则化最小二乘来预测 miRNA 和疾病之间可能的关联。我们使用 GATs 从已知 miRNA-疾病关联的初始输入特征,miRNA 内部相似性和疾病内部相似性中学习每层 miRNA 和疾病的嵌入。然后,我们基于高斯互作谱(GIP)与学习到的嵌入计算 miRNA 和疾病的核矩阵。进一步将各层的核矩阵和初始相似性利用注意力机制融合。双拉普拉斯正则化最小二乘最终与融合 miRNA 和疾病核应用于新的 miRNA 与疾病的关联预测。通过5折交叉验证与的6种最新方法相比,我们的方法 MKGAT 获得最高的 AUROC 值为0.9627,AUPR 值为0.7372。我们使用 MKGAT 来预测三种癌症的相关 miRNA,发现三种疾病的前50个预测结果都被现有的数据库所证实。优异的性能表明,MKGAT 将是一个有用的计算工具,以揭示疾病相关的 miRNA。
关键词: miRNA-疾病关联,图注意力网络,多核融合,双拉普拉斯正则化最小二乘
目录
2.材料和方法(Materials and methods)
2.1.1.Known human miRNA–disease associations
2.1.2.miRNA functional similarity
2.1.3.Disease semantic similarity
2.1.4.GIP kernel similarity for diseases and miRNAs
2.1.5.Integrated similarities for miRNAs and diseases
2.1.6.miRNA-disease bipartite network
2.2.1.用于特征提取的 GATs(用于特征提取的 GATs)
1.引言(Introduction)
在这项研究中,我们提出了一个计算框架 MKGAT,它结合了 GATs [45]和双拉普拉斯正则化最小二乘来预测潜在的 miRNA 疾病关联。首先,输入特征是基于已知的 miRNA-疾病关联、 miRNA 内部相似性和疾病内部相似性构建的。GATs 被用来研究在每一层上 miRNA 和疾病的嵌入。然后,基于高斯互作谱(GIP)计算每一层的 miRNA 和疾病嵌入的核矩阵,并利用注意力机制将每一层的核矩阵和初始相似性融合。最后,在结合 miRNA 和疾病核的空间用双拉普拉斯正则化最小二乘预测新的 miRNA 疾病关联。五折交叉验证显示,MKGAT 的 ROC曲线曲线下面积(AUROC)为0.9627,精确召回曲线下面积(AUPR)为0.7372,优于六种最先进的预测方法。对三种癌症的病例研究显示,所有前50位的预测都得到了已建立的数据库的支持,这进一步证明了 MKGAT 在检测疾病相关的 miRNA 方面的有效性。
2.材料和方法(Materials and methods)
2.1.基准数据集(Benchmark datasets)
2.1.1.Known human miRNA–disease associations
研究中使用的数据集从参考文献[26]下载,其中 Chen 等人从 HMDD v2.0收集了495个 miRNA,383个疾病和5430个实验验证的 miRNA 疾病关联[46]。我们使用 ![]() 和
和 ![]() 分别表示 miRNA 和疾病的数量,并且
分别表示 miRNA 和疾病的数量,并且 ![]() 来描述 miRNA-疾病关联的邻接矩阵,其中
来描述 miRNA-疾病关联的邻接矩阵,其中 ![]() (= 495)表示行数(miRNA) ,
(= 495)表示行数(miRNA) ,![]() (= 383)表示列数(疾病)。如果 miRNA m(i) 和疾病 d(j) 具有已知的关联,则矩阵相应位置的
(= 383)表示列数(疾病)。如果 miRNA m(i) 和疾病 d(j) 具有已知的关联,则矩阵相应位置的 ![]() 的值设置为1,否则为0。
的值设置为1,否则为0。
2.1.2.miRNA functional similarity
Wang 等[47]基于表型相似的疾病更可能与功能相似的 miRNA 相关的假设,提供了一种 miRNA 功能相似性计算方法。我们从他们的研究 https://www.cuilab.cn/files/images/cuilab/misim.zip 下载了 miRNA 的功能相似性。我们构建了一个矩阵 FS 来描述两个 miRNA 之间的功能相似性,其中![]() 表示 miRNA
表示 miRNA![]() 和
和![]() 之间的 miRNA 功能相似性评分。
之间的 miRNA 功能相似性评分。
2.1.3.Disease semantic similarity
我们使用 MeSH 术语将每种疾病描述为一种有向无环图(DAG)。具体而言,我们使用 ![]() 制定疾病 di,其中
制定疾病 di,其中 ![]() 表示由节点 di 及其祖先节点组成的节点集,
表示由节点 di 及其祖先节点组成的节点集,![]() 表示包含从祖先到子节点的直接链接的相应边集。根据参考文献[47] ,我们计算疾病 dt 对 di 的语义贡献如下:
表示包含从祖先到子节点的直接链接的相应边集。根据参考文献[47] ,我们计算疾病 dt 对 di 的语义贡献如下:
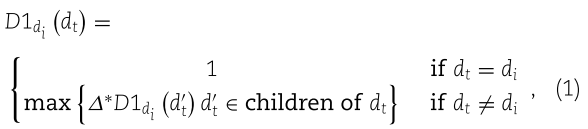
![]() 其中表示语义贡献衰减因子,在我们的研究中设置为0.5。疾病 di 的语义值可根据祖先疾病和疾病 di 本身的贡献计算如下:
其中表示语义贡献衰减因子,在我们的研究中设置为0.5。疾病 di 的语义值可根据祖先疾病和疾病 di 本身的贡献计算如下:
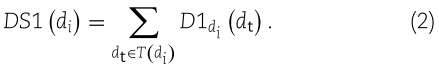
疾病 di 和 dj 之间的语义相似度 ![]() 由以下公式定义:
由以下公式定义:
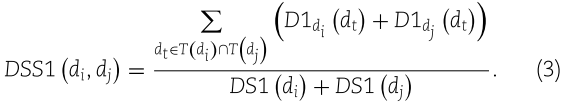
同时,陈等[26]提出用另一种方法计算疾病 dt 对 di 的语义贡献

相应地,疾病 di 的语义值和疾病 di 与 dj 之间的语义相似度可以分别由方程(5)和(6)计算出来,
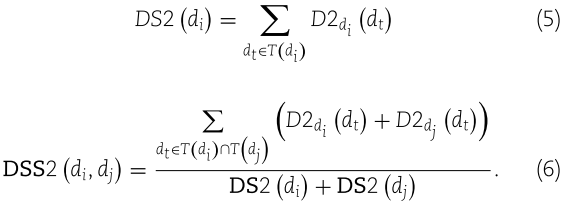
在本研究中,我们通过结合这两种疾病的语义相似度来计算最终的疾病语义相似度,并且可以用方程(7)来计算两种疾病之间的疾病语义相似度 SS (di,dj)
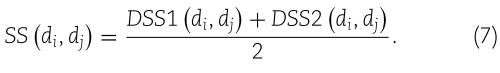
2.1.4.GIP kernel similarity for diseases and miRNAs
类似于参考文献[48] ,构建了一个二元载体![]() 来记录 miRNA m (i)与所有疾病之间的关联。如果两者之间存在实验支持的关联,则
来记录 miRNA m (i)与所有疾病之间的关联。如果两者之间存在实验支持的关联,则![]() 的对应值设置为1,否则设置为0。然后可以计算 miRNA mi 和 mj 之间的 GIP 核相似性
的对应值设置为1,否则设置为0。然后可以计算 miRNA mi 和 mj 之间的 GIP 核相似性 ![]() 如下:
如下:
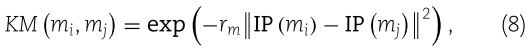
其中 ![]() 表示标准化的内核带宽,并从方程(9)中获得如下:
表示标准化的内核带宽,并从方程(9)中获得如下:

其中 Nm 表示所有 miRNA 的数量,![]() 是原始带宽,在我们的研究中设置为1。类似地,两种疾病 di 和 dj 之间的 GIP 核相似度
是原始带宽,在我们的研究中设置为1。类似地,两种疾病 di 和 dj 之间的 GIP 核相似度 ![]() 可以用以下两个方程计算:
可以用以下两个方程计算:

其中 ![]() 表示标准化的核带宽,
表示标准化的核带宽,![]() 表示所有疾病的数目,
表示所有疾病的数目,![]() 被设置为1。
被设置为1。
2.1.5.Integrated similarities for miRNAs and diseases
考虑到并非所有 miRNA 对都具有功能相似性,miRNA mi 和 mj 之间的整合相似性 ![]() 计算如下:
计算如下:
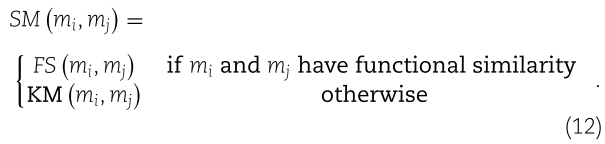
同样,疾病 di 和 dj 之间的整合相似性 ![]() 计算如下:
计算如下:
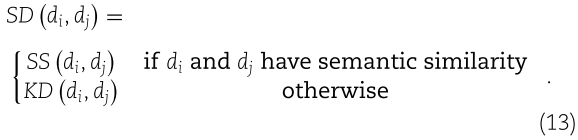
这种相似性整合策略已应用于参考文献[23,26,49]中的 miRNA-疾病关联推断。
2.1.6.miRNA-disease bipartite network
MiRNA-疾病二分网络 G 是由邻接矩阵![]() 及其转置
及其转置 ![]() 定义的
定义的
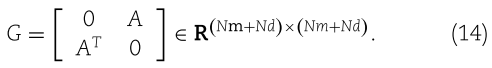
2.2.方法架构(Method architecture)
在本节中,我们将介绍用于 miRNA 疾病关联预测的 MKGAT 的体系结构。MKGAT 的工作流程如图1所示。

2.2.1.用于特征提取的 GATs(用于特征提取的 GATs)
作为一种新的神经网络结构,GATs [45]被应用于我们的研究中,以提取 miRNA 和疾病特征。具体而言,鉴于上述二分网络的邻接矩阵 G,GATs的定义如下:

其中 ![]() 是节点的 l 层嵌入,l = 1,... ,L,σ (•)是非线性激活函数(ReLU) ,GAT 表示一个单一的图注意层,整个 L 层结构由多个图注意层叠加而成。初始输入是一组节点特征
是节点的 l 层嵌入,l = 1,... ,L,σ (•)是非线性激活函数(ReLU) ,GAT 表示一个单一的图注意层,整个 L 层结构由多个图注意层叠加而成。初始输入是一组节点特征 ![]() ,其中 N 是节点数,F 是每个节点的特征数。该层产生一组新的节点特征
,其中 N 是节点数,F 是每个节点的特征数。该层产生一组新的节点特征 ![]() ,通过对每个节点应用加权矩阵
,通过对每个节点应用加权矩阵 ![]()
![]() ,使用可学习的线性映射将输入特征转换为更高层次的特征。然后我们计算注意力系数
,使用可学习的线性映射将输入特征转换为更高层次的特征。然后我们计算注意力系数
![]()
经过softmax激活函数归一化后,我们得到的系数是

将方程(16)代入方程(17) ,注意机制的系数可以表示如下:
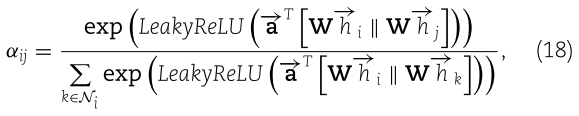
其中 α 表示注意力系数,![]() 表示参数化权重向量,LeakyReLU 表示激活函数(负斜率为0.2) ,T 表示矩阵转置,| | 表示连接运算,Ni 表示节点 i 的邻居。在计算归一化注意系数后,每个节点的最终输出特征可以计算为
表示参数化权重向量,LeakyReLU 表示激活函数(负斜率为0.2) ,T 表示矩阵转置,| | 表示连接运算,Ni 表示节点 i 的邻居。在计算归一化注意系数后,每个节点的最终输出特征可以计算为
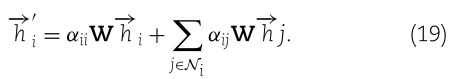
在我们的研究中,我们构造了第一层的初始嵌入 ![]() 如下:
如下:
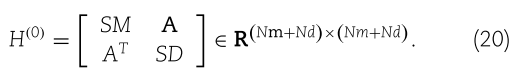
2.2.2.Kernel combination
多层 GAT 模型计算不同层的嵌入,每一层的嵌入表示不同的图结构信息。我们通过将每一层分别作为不同的特征向量嵌入来计算多核矩阵。表示每一层的嵌入为 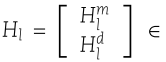
![]() ,其中
,其中 ![]() 是 miRNA 在第 l 层的嵌入,
是 miRNA 在第 l 层的嵌入,![]() 是疾病在第 l 层的嵌入。我们使用 GIP 计算 miRNA 和疾病的每一层嵌入的核矩阵如下:
是疾病在第 l 层的嵌入。我们使用 GIP 计算 miRNA 和疾病的每一层嵌入的核矩阵如下:
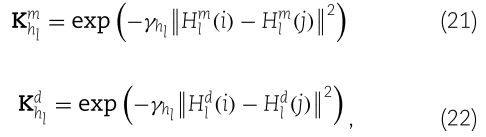
其中 ![]() 表示 miRNA 和疾病嵌入的第 l 层中的第 l 行,
表示 miRNA 和疾病嵌入的第 l 层中的第 l 行,![]() 表示相应的宽度。
表示相应的宽度。
由于不同层的不同嵌入的贡献是不一致的,嵌入计算的核表示不同视图节点之间的相似性。对 miRNA 空间 ![]()
![]()
![]() 是初始相似矩阵 SM,疾病空间
是初始相似矩阵 SM,疾病空间 ![]() ,
,![]() 是初始相似矩阵 SD。利用注意机制组合多个核矩阵(分别在两个空间中) ,得到最终的组合核
是初始相似矩阵 SD。利用注意机制组合多个核矩阵(分别在两个空间中) ,得到最终的组合核
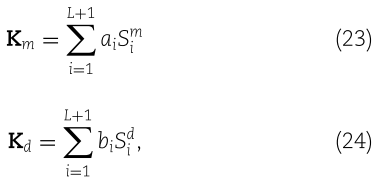
其中![]() 是 miRNA 和疾病核的第 i 核,
是 miRNA 和疾病核的第 i 核,![]() 是每个核对应的注意因子,L 是层数。
是每个核对应的注意因子,L 是层数。
2.2.3.预测的双拉普拉斯正则最小二乘法
50. Belkin M, Niyogi P, Sindhwani V. Manifold regularization: a
geometric framework for learning from Labeled and Unlabeled
examples. J Mach Learn Res 2006;7:2399–434.
我们应用双拉普拉斯正则化最小二乘[50]于两个特征空间的联合核矩阵来预测 miRNA 与疾病之间的潜在关联。损失函数定义为
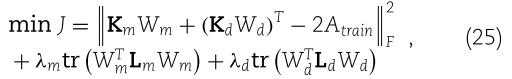
其中 ![]() 是 Frobenius 范数,
是 Frobenius 范数,![]() 是训练集中 miRNA-疾病关联的邻接矩阵,
是训练集中 miRNA-疾病关联的邻接矩阵,![]() 是可训练矩阵,
是可训练矩阵,![]() 和
和 ![]() 是两个特征空间中的组合核,参数 λm 和 λd 是正则项的系数。
是两个特征空间中的组合核,参数 λm 和 λd 是正则项的系数。
![]()
![]() 是拉普拉斯正则矩阵,定义如下:
是拉普拉斯正则矩阵,定义如下:
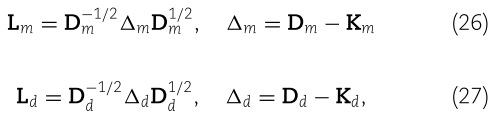
其中 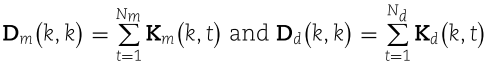 是对角矩阵。最后的 miRNA-疾病关联预测如下:
是对角矩阵。最后的 miRNA-疾病关联预测如下:
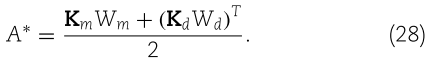
2.2.4.参数优化
我们使用 Adam 优化器[51]来优化 GAT 中的参数和用于核融合的注意因子。对于双拉普拉斯正则最小二乘的参数,我们通过直接计算偏导数得到迭代函数。为了优化参数 Wm,我们将参数 Wd 固定为常数,然后计算损失函数相对于 Wm 的偏导数如下:

3.结果
3.1.实验设置
3.2.不同核对预测性能的影响
3.3.与其他方法的性能比较
3.4.案例研究
4.结论
生命科学的最新进展表明,miRNA 在发育和生理调控中起着关键作用。因此,miRNA 正在成为疾病诊断的重要生物标志物。预测疾病相关 miRNA 的计算工作是生物医学实验的一个极好的替代方案。在本文中,我们提出了一个计算框架 MKGAT 来发现 miRNA 与疾病之间的关联。包括交叉验证和病例研究在内的综合实验显示了 MKGAT 在揭示疾病相关 miRNA 方面的有效性和优越性。
我们的框架 MKGAT 主要由两部分组成。在第一部分中,我们使用了一种注意机制来提取特征。实验表明,利用该机制可以产生更可靠的推理信息。另一部分是用于预测的对偶拉普拉斯正则最小二乘法。作为一种实体和半监督的方法,我们的双 Laplacian 正则化最小二乘方法充分利用了来自 miRNA 侧和疾病侧的信息进行预测。与使用分层 GAT 进行推断的方法 HGANMDA [44]相比,我们的模型 MKGAT 和 HGANMDA 之间有三个主要差异。首先,MKGAT 使用 GIP 来计算核矩阵。其次,MKGAT 采用注意机制对多个核矩阵进行组合。第三,MKGAT 使用不同的策略进行最终预测。 与有监督的 miRNA 疾病关联预测方法相比,我们的框架不需要阴性样本进行推断。正如我们在引言部分所述,用于 miRNA-疾病关联预测的阴性样本的数据很难收集,随机选择的阴性样本将输出不太令人满意的结果。
值得注意的是,我们的方法在很大程度上依赖于相似性度量来进行推理。正如之前的研究[59]所述,整合适当的特征进行相似性计算是一项具有挑战性的任务,因为在生物医学科学的数据可用性方面存在缺陷。例如,疾病可能不包括在 MeSH 数据库中。此外,正如我们在引言部分所述,MKGAT 中的参数调优和优化是一个棘手的过程。这是深度学习方法中需要解决的一个常见问题。最后,作为人类疾病进展的调节因子,miRNA 可以上调或下调基因表达。然而,在本研究中我们没有区分调节模式。这将是我们未来研究的一个新方向。
