A learning-based framework for miRNA-disease association identification using neural networks
源代码:GitHub - Issingjessica/MDA-CNN: this is the source code and data

摘要
动机:微小 RNA (miRNA)是一种非编码 RNA,在许多生物学过程中起着重要作用。许多研究表明,miRNA 与人类疾病有关,这表明 miRNA 可能是各种疾病的潜在生物标志物。因此,揭示 miRNA 与疾病/表型之间的关系具有重要意义。
结果:我们提出了一个新的基于学习的框架,MDA-CNN,用于 miRNA 疾病的关联鉴定。该模型首先基于包括疾病相似性网络、 miRNA 相似性网络和蛋白质-蛋白质相互作用网络在内的三层网络捕获疾病与 miRNA 之间的相互作用特征。然后,采用自动编码器对每对 miRNA 和疾病的基本特征组合进行自动识别。最后,以简化的特征表示作为输入,使用一个卷积神经网络来预测最终的标签。评估结果表明,所提出的框架在 miRNA 疾病关联预测和 miRNA 表型关联预测两个任务上大大优于一些最先进的方法。
目录
2.1.基于网络的特征提取(Network-based feature extraction)
2.1.1.关联得分计算(Association score calculation)
2.1.2.特征表示(Feature representation)
2.2.基于自动编码器的特征选择(Auto-encoder-based feature selection)
2.3.基于卷积神经网络的关联预测(Convolutional neural network-based association prediction)
3.3.Performance evaluation on predicting miRNA-disease associations
3.4.Performance evaluation on predicting miRNA-phenotype associations
3.5.Effects of MDA-CNN components
1.引言
在本文中,我们提出了一个新的基于学习的框架,MDA-CNN,以确定之间的联系,一对 miRNA 和疾病。以下是四个主要贡献:
我们为 miRNA 疾病关联预测任务引入了一个基于学习的框架,该框架包含三个组成部分,即基于网络的特征提取器,基于自动编码器的特征选择器和基于 CNN 的关联预测器。
为了更好地表示 miRNA 与疾病之间的相关性,我们构建了一个三层网络,中间增加了一个基因层。在此基础上,提出了一种新的基于回归模型的特征表示方法。
我们使用深层 CNN 架构来处理前一步产生的特征向量,以确定 miRNA-疾病对的最终标签。
评估结果显示,MDA-CNN 在 miRNA 疾病和 miRNA 表型关联鉴定方面优于一些最先进的方法。
2.材料及方法
我们提出了一种新的算法称为 MDA-CNN 来预测 miRNA 疾病的关联。MDA-CNN 的框架如图1所示,它包含三个步骤。首先,给定一个三层网络(图1a) ,我们应用回归模型来计算 疾病 - 基因 和 miRNA - 基因 关联评分,并根据这些关联评分生成疾病和 miRNA 对的特征向量。其次,给定一对 miRNA 和疾病,相应的特征向量通过基于自动编码器的模型来获得低维表示(图1b)。第三,基于最后一步获得的表达载体(图1c) ,构建了一个深层的 CNN 结构来预测 miRNA 与疾病之间的关联。

2.1.基于网络的特征提取(Network-based feature extraction)
众所周知,miRNA 通过调控转录后基因表达与许多疾病有关。 在这项工作中,我们添加了基因层网络作为桥梁,以提取miRNA-疾病对之间的相互作用特征。我们利用人类基因的 PPI 网络作为基因层网络。MiRNA 疾病对的特征提取包括两个步骤: (i)基于 PPI 网络和疾病(miRNA)网络计算疾病(miRNA)和基因之间的关联评分; (ii)为 miRNA-疾病对生成特征向量。对于每个 miRNA 疾病对,其特征向量是疾病向量和 miRNA 向量的串联。疾病(miRNA)特征向量中的元素代表了疾病(miRNA)与 PPI 网络中每个基因之间的关系。我们计算一个关联得分来测量疾病(miRNA)和基因之间的关系,而不是表示疾病(miRNA)和基因是否相关的二进制值。
2.1.1.关联得分计算(Association score calculation)
接下来,我们以疾病和基因层为例来说明我们的算法。miRNA 和基因之间的关联得分可以类似地计算出来。
设 ![]() 和
和 ![]() 分别是一个疾病网络和一个基因关联网络。
分别是一个疾病网络和一个基因关联网络。![]() 和
和 ![]() 分别代表
分别代表 ![]() 和
和 ![]() 所涉及的一系列疾病和基因。
所涉及的一系列疾病和基因。![]() 是
是 ![]() 和
和 ![]() 之间的一组疾病基因关联性。受 Wu 等人(2008)的启发,疾病 d 和基因 g 之间的关联得分可以用
之间的一组疾病基因关联性。受 Wu 等人(2008)的启发,疾病 d 和基因 g 之间的关联得分可以用 ![]() 和
和 ![]() 的 Pearson 相关系数来衡量
的 Pearson 相关系数来衡量

其中 ![]() 是 d 和
是 d 和 ![]() 中每种疾病之间相似性得分的向量,
中每种疾病之间相似性得分的向量,![]()
![]() 是 g 和
是 g 和![]() 中每种疾病之间的亲密度得分的向量,
中每种疾病之间的亲密度得分的向量,![]() ,
,![]() 分别代表协方差和标准差。
分别代表协方差和标准差。
给定 ![]() 和
和 ![]() ,基因 g 和疾病 d 之间的亲密度得分可以定义如下:
,基因 g 和疾病 d 之间的亲密度得分可以定义如下:

其中 ![]() 是与 d 相关的一组基因;
是与 d 相关的一组基因; ![]() 是
是 ![]() 中
中 ![]() 和
和 ![]() 之间最短路径的平方。
之间最短路径的平方。
我们不使用基于路径距离的相似性,而是使用回归模型来计算两种疾病 di 和 dj 之间的相似性。该模型能够考虑基因对不同疾病的重要性。该模型的定义如下:

![]() 是这个线性回归模型的回归系数,
是这个线性回归模型的回归系数,![]() 是一组与
是一组与 ![]() 相关的基因,而
相关的基因,而  是每种疾病的偏差常数。
是每种疾病的偏差常数。 代表了
代表了 ![]() 对
对 ![]() 的重要性。该回归模型的基本思想是通过相关基因测定两种疾病的相似性。给定
的重要性。该回归模型的基本思想是通过相关基因测定两种疾病的相似性。给定 ![]() ,这个线性回归模型可以训练和用来计算疾病的相似性。请注意,
,这个线性回归模型可以训练和用来计算疾病的相似性。请注意,![]() 与
与 ![]() 不同。
不同。
2.1.2.特征表示(Feature representation)
特征表示是应用机器学习算法的关键步骤。 然而,大多数现有的研究使用关联分数来连接疾病和 miRNA,这并不是为了表现疾病和 miRNA 之间复杂的相互作用而设计的。通过添加基因层,我们可以产生一个向量来表示 miRNA-疾病对的特征。
给定一个疾病 d,我们根据方程(1)计算 d 与基因层中涉及的每个基因之间的关联得分。在此之后,d 的特征向量可以生成为
![]()
其中 ![]() 代表
代表 ![]() 涉及的一个基因,
涉及的一个基因,![]() 是
是 ![]() 涉及的基因数。为了减少
涉及的基因数。为了减少 ![]() 中极端值(异常值)的影响,我们对
中极端值(异常值)的影响,我们对 ![]() 应用 softmax 标准化(Grover and Leskovec,2016)。具体来说,归一化向量
应用 softmax 标准化(Grover and Leskovec,2016)。具体来说,归一化向量 ![]() 表示如下:
表示如下:

同样,给定一个 miRNA ![]() ,可以生成以下向量
,可以生成以下向量 ![]() :
:
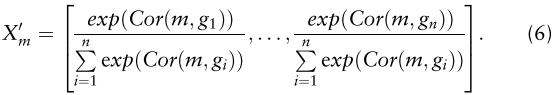
对于 miRNA-疾病对,我们连接 ![]() 和
和 ![]() 作为特征表示的载体。
作为特征表示的载体。
2.2.基于自动编码器的特征选择(Auto-encoder-based feature selection)
前一步生成的向量长度(例如 ![]() 和
和 ![]() 的级联)是
的级联)是 ![]() 的两倍,它非常大而且有噪声。因此,我们应用自动编码器来识别基本特征组合,并自动降低每对 miRNA 和疾病的特征向量的维数。
的两倍,它非常大而且有噪声。因此,我们应用自动编码器来识别基本特征组合,并自动降低每对 miRNA 和疾病的特征向量的维数。
自动编码器用于下游机器学习任务的降维,例如分类、可视化、通信和高维数据的存储(Chicco et al。 ,2014)。与广泛使用的主成分分析(PCA)方法不同,自动编码器是 PCA 的非线性推广,它使用自适应“编码器”网络将高维数据转换为低维编码,并使用类似的“解码器”网络从低维编码中恢复数据。然后使用低维代码作为原始数据的压缩表示。自动编码器的细节在补充文档中有描述。
在我们的实验中,向量 ![]() 和
和 ![]() 在进入自动编码器模型之前被串联。设 n 是涉及网络
在进入自动编码器模型之前被串联。设 n 是涉及网络 ![]() 的基因数。输入的原始尺寸为2n。在我们的模型中,我们使用均方误差(MSE)(Wax 和 Ziv,1977)作为损失函数。使用 sigmoid 激活函数和 Adam 算法来优化 MSE 损失。我们的自动编码器网络是由反向传播(BP)算法训练(Rumelhart 等,1988)。
的基因数。输入的原始尺寸为2n。在我们的模型中,我们使用均方误差(MSE)(Wax 和 Ziv,1977)作为损失函数。使用 sigmoid 激活函数和 Adam 算法来优化 MSE 损失。我们的自动编码器网络是由反向传播(BP)算法训练(Rumelhart 等,1988)。
2.3.基于卷积神经网络的关联预测(Convolutional neural network-based association prediction)
20世纪80年代末,Lecun (Lecun 等,1989)提出了 CNN,其在图像分类(Krizhevsky 等,2012) ,句子分类(Kim,2014)和结构图数据分类任务(Atwood 和 Towsley,2016)。在这项工作中,我们还选择了卷积神经网络作为监督式学习模型,以了解特征的最佳组合,并预测给定的 miRNA-疾病对的最终标签。所提出的模型的结构如图2所示。我们的模型包括以下几个层次: 卷积和激活层,最大池化层,全连接层和softmax层。卷积层和校正线性单元[ ReLU,(Nair and Hinton,2010)]激活层用于从输入中提取特征,这是维度减化步骤的输出(见第2.2节)。池化层层用于降维。最后的完全连接层和 softmax 层用于分类任务。

卷积层负责学习输入的子空间特征。模型的卷积层由四个卷积核组成。4 * 1权重向量与长度为 L 的输入向量卷积。在卷积之后,对于每个核,我们可以得到一个特征映射 C (从输入中提取的特定特征) ,它是一个具有长度![]() 的向量。特征映射 C 由以下方程式提取:
的向量。特征映射 C 由以下方程式提取:

其中 ![]() ,X 是输入向量,
,X 是输入向量,![]() 是权重向量,初始化为截断的正态分布,平均值为0,标准差为0.1。
是权重向量,初始化为截断的正态分布,平均值为0,标准差为0.1。![]() 高表明核能很好地捕捉输入子区域的特征。然后
高表明核能很好地捕捉输入子区域的特征。然后![]() 通过一个 ReLU 函数
通过一个 ReLU 函数 ![]() ,忽略负输出,并传播来自前一层的正输出。尽管存在各种非线性,但由于其计算效率,稀疏性和消失梯度的可能性降低,ReLU 激活是最受欢迎的(Krizhevsky 等,2012; Lecun 等,2015)。
,忽略负输出,并传播来自前一层的正输出。尽管存在各种非线性,但由于其计算效率,稀疏性和消失梯度的可能性降低,ReLU 激活是最受欢迎的(Krizhevsky 等,2012; Lecun 等,2015)。
最大池化层用于对卷积层之后的潜在表征进行下采样。它取卷积层输出的非重叠子区域(即池大小为4)上的最大值,并在每个特征映射的邻域上输出最重要的特征。给定一个输入序列![]() ,池层的输出如下所示:
,池层的输出如下所示:

![]()
卷积层和最大池化层可以从输入向量中提取重要特征。然后,将所有核的输出连接到一个向量,并提供给全连接层。
最后两层是一个完全连接的层和一个softmax层。在全连接层中有50个隐藏单元。池化层的输出为 ![]() ,其中 n 是池层的级联输出的长度。完全连接层的输出是:
,其中 n 是池层的级联输出的长度。完全连接层的输出是: ![]() ,其中
,其中 ![]() 是权重矩阵,f 是 ReLU 激活。最终的 softmax 层用于分类任务。
是权重矩阵,f 是 ReLU 激活。最终的 softmax 层用于分类任务。
3.结果
3.1.Experiment setup
我们在两个任务上评估我们的模型,即 miRNA 疾病关联预测和 miRNA 表型关联预测。两个任务中使用的数据集是独立的,没有重叠。在 miRNA 疾病关联预测的测试数据集中,阳性集从 HMDDv2.0 获得(Li 等,2013b)(http://www。Cn/hmdd).HMDD 是一个手动收集的数据库与 miRNA 疾病的关联实验支持的证据。由于负样本没有可用的数据集,所以我们随机生成一个与正样本大小相同的负样本集。对于 miRNA 表型关联,从 miRwalk2.0数据库(Dweep 和 Gretz,2015)( http://zmf.umm.uni-heidelberg.de/ apps/zmf/miRWalk2/)获得一组经过验证的 miRNA 表型关联。MiRWalk 包括经过验证的 miRNA 与表型之间的相互作用。我们还随机生成一个与正集大小相同的负集。在这两个任务中,我们使用10倍交叉验证(Kohavi 等,1995)。评估指标包括 ROC曲线曲线下的面积(AUROC)、精确召回曲线下的面积(AUPR)、精确度、召回和 f 1分数。
3.2.Data description
对于 miRNA-疾病关联预测,我们需要对相同类型的元素,即疾病,基因和 miRNA 有三个相似性网络。我们从 You 等人(2017)获得疾病相似性网络和 miRNA 相似性网络(http://www.escience.cn/system/file?%20fileId=84394)。我们利用来自人类蛋白质参考数据库(HPRD)的人类基因的蛋白质网络(宝林和波,2007)( Human Protein Reference Database )。涉及不同网络的关联是疾病基因和 miRNA 基因的关联。疾病与基因的关联来自 DisGeNET 数据库(Pi ~ nero et al。 ,2016)( http://www.disgenet.org/web/DisGeNET/menu ) ,只有手工策划的疾病与基因的关联被保留。miRNA 基因关联获自 miRWalk2.0数据库(Dweep 和 Gretz,2015)(http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/).在计算过程中,我们去除了那些与疾病或 miRNAs 无关的基因。
3.3.Performance evaluation on predicting miRNA-disease associations
3.4.Performance evaluation on predicting miRNA-phenotype associations
3.5.Effects of MDA-CNN components
3.6.Case study
4.结论
最近,研究人员开始着重于通过计算工具识别 miRNA 疾病的相关性。在本文中,我们提出了一个基于学习的框架,命名为 MDA-CNN 来鉴定 miRNA 疾病/表型的关联。我们首先基于一个三层网络提取 miRNA 和疾病/表型的特征。然后,提出了一种基于自动编码器的特征选择模型。使用这个特征表示,我们提出了一个卷积神经网络结构,用于预测 miRNA 疾病/表型的关联。为了说明 MDA-CNN 的优点,我们将其与三种最先进的方法进行了比较。对 miRNA 疾病和 miRNA 表型相关性的实验表明,MDA-CNN 比现有的方法表现得更好,表明所提出的基于学习的框架设计得当。此外,对肺癌和心力衰竭的病例研究表明,MDA-CNN 可用于预测 miRNA 疾病的相关性。在未来,我们将开发一个网络服务器,包括更多类型的数据集,以方便使用 MDA-CNN。