NMCMDA: neural multicategory MiRNA–disease association prediction
![]()
![]()
目录
3.1.miRNA与疾病的多种关联(Multiple categories of MiRNA–disease associations)
3.3.疾病-疾病相似性(Disease–disease similarity)
4.2.miRNA与疾病潜在特征的编码器(Encoders for latent feature of MiRNA and disease)
4.4.端到端学习模型(End-to-end learning models)
5.结果和讨论(Results and discussion)
5.2.各种编码器-解码器组合的性能分析(Performance analysis of various encoder–decoder combinations)
5.4.与现有工作的比较(Comparisons with existing work)
1.摘要(Abstract)
动机:越来越多的证据表明,miRNAs的失调通过各种潜在的机制导致疾病。 因此,预测microRNAs(miRNAs)与疾病的多类关联对于研究miRNAs在疾病中的作用具有重要意义。 此外,与传统的耗时昂贵的生物学实验相比,预测多基因miRNA与疾病关联的计算方法节省了时间和成本效益,是我们迫切需要的。
结果:我们提出了一种新的基于数据驱动的端到端学习的神经多类miRNA疾病关联预测方法(NMCMDA)来预测多类miRNA疾病关联。 NMCMDA由两个主要部分组成:(一)编码器直接作用于miRNA-疾病异构网络,利用图神经网络分别学习miRNA和疾病的潜在表征。 (ii)解码器以学习到的潜在表征作为输入,产生miRNA-疾病关联分数。 针对NMCMDA提出了各种编码器和解码器。 最后,采用关系图卷积网络编码器和神经多关系解码器的NMCMDA(NMR-RGCN)获得了最佳的预测性能。 我们在三个实验数据集上比较了NMCMDA和其他基线。 实验结果表明,NMR-RGCN在Top-1精确度、Top-1查全率和Top-1 F1等方面明显优于TDRC方法。 此外,我们还对两种高危疾病(乳腺癌和肺癌)进行了案例研究,并根据HMDD V3.2的所有已知数据对Top-10 miRNA疾病类别关联进行了预测和验证,进一步验证了该方法的有效性和可行性。
关键词:microRNA; 疾病; 多类miRNA与疾病的关联; 关系图卷积网络; 神经多关系解码器
2.引言(Introduction)
MiRNA 是一种小的内源性非编码 RNA(长度为 21-24 个核苷酸),在多种生物过程中起着至关重要的作用 [1-3]。 miRNA 及其靶 mRNA 的功能障碍可能导致各种人类疾病 [4]。如肺癌中let-7的表达水平明显降低,证实miRNA与肿瘤的发生密切相关[5]; mir-107的异常表达可能影响BACE1(β-分泌酶1)的活性而引起阿尔茨海默病[6]。 因此,疾病相关 miRNA 的鉴定有助于疾病的病理学研究和疾病生物标志物的检测[7-11]。由于使用生物学实验鉴定 miRNA 与疾病之间的关联是耗时和昂贵的[12] ,在过去几年中,已经开发了许多计算方法[13-25]来确定 miRNA 与疾病之间的潜在关联(以下简称 MDA)。
现有的大多数研究工作,如[13-25] ,主要集中在二元关联预测(即仅预测 miRNA-疾病关联的存在) ,并确实取得了良好的结果。然而,越来越多的证据表明,miRNAs的调控异常通过多种潜在机制引起疾病[26-29]。 例如,表观遗传学改变可能导致miRNAs的异常表达而引起疾病:启动子甲基化降低mIR-17 ̄92簇的表达水平,导致支气管肺发育不良[30]; miRNAs与靶点的相互作用与多种疾病有关:miR-101通过靶向CXCL12抑制成纤维细胞与癌细胞的相互作用,从而影响肺癌细胞的增殖[31]。 此外,同一miRNA可能通过不同的关联机制与同一疾病发生关联。 如前面提到的miR-101通过靶向CXCL12作用于肺癌,同时miR-101也通过抑制肺癌细胞DNA甲基化而抑制肺癌[32]。 上述方法无法确定miRNA与疾病关联的具体类别。 因此,识别多种类型的miRNA与疾病的关联,不仅可以提供更详细的miRNA与疾病之间的潜在关联,而且可以在miRNA水平上进一步加深我们对疾病分子基础的理解。
在过去的几年里,只有很少的研究工作致力于确定多种类型的miRNA与疾病的关联multiple categories of miRNA–disease associations(MCMDA)。 Chen等人[33]首先研究MCMDA问题。 在他们的研究中,发展了用于多种类型miRNA-疾病关联预测的限制性玻尔兹曼机模型(RBMMMDA)来预测四种不同类型的miRNA-疾病关联。 基于该模型,不仅可以得到新的miRNA与疾病的关联,而且可以得到相应的关联类型。 Zhang等人[34]提出了一种称为基于网络的标签传播算法(NLPMMDA)的半监督模型,该模型通过从异构网络中获得的互信息来推断多种类型的miRNA与疾病的关联。 注意,上述两项研究基于2013年发布的人类microRNA疾病数据库2.0[26](以下简称HMDD V2.0)开发了他们的方法。
最近发布的人类microRNA疾病数据库3.0[27](最新版本为HMDD V3.2)提供了6个广义的关联类别,包括分别从遗传学、表观遗传学、循环miRNAs、组织、miRNA-靶点和其他相互作用的证据出发的miRNA-疾病关联。 这些关联涵盖了20个不同类别的详细关联证据代码。 HMDD V3.2在miRNA、疾病条目、miRNA-疾病关联条目方面显著扩展了HMDD V2.0,并提供了更多的关联类别信息。 这为开发新的数据驱动的MCMDA方法提供了机会。
最近,针对HMDD V3.2数据集,Huang等人[35]将多类miRNA-疾病关联表示为一张量,并将多类miRNA-疾病关联预测表述为一张量补全任务。 他们提出了一种新的基于张量分解的关系约束张量分解模型(TDRC)来解决MCMDA,该模型将miRNA与miRNA相似度和疾病与疾病相似度信息作为分解约束。 实验结果显示TDRC显著优于其他基线[33-35]。
尽管 TDRC 和其他方法对 MCMDA 有效,但目前的研究结果仍然存在一些局限性。首先,miRNA-疾病关联预测依赖于功能相似的 miRNA 通常与表型相似的疾病相关的假设,反之亦然。因此,miRNA 和疾病信息的相似性对于准确预测至关重要。然而,RBMMMDA [33] 仅仅利用了已知的多类别 miRNA-疾病关联,并没有考虑 miRNA(疾病)之间信息的相似性。这限制了 RBMMMDA 的预测性能。此外,虽然 TDRC [35] 以 miRNA-miRNA 相似性和疾病-疾病相似性信息作为分解约束,但张量分解的技术框架不能轻易使用其他来源的 miRNA 和疾病特征信息。
第二,NLPMMDA[34]将预测一类miRNA与疾病的关联作为一项独立的任务,因此忽略了关联类别之间的相关性。 然而,不同的关联类别可能彼此高度关联。 TDRC[35]利用张量分解,通过张量乘法来捕捉miRNAs与疾病和关联类别之间复杂的多线性关系,克服了上述局限性。 然而,TDRC本质上是一种多线性方法,可能不足以捕捉miRNAs特征与疾病之间复杂而非线性的相互作用。
为了克服现有MCMDA方法的上述局限性,我们提出了一种新的基于数据驱动的端到端学习的神经多类别miRNA疾病关联预测方法(NMCMDA)来解决MCMDA问题。 NMCMDA由两个主要部分组成:编码器直接作用于miRNA-疾病异构网络,利用图神经网络分别学习miRNA和疾病的潜在表征。 解码器以学习到的潜在表征作为输入,产生miRNA-疾病关联分数。 本研究提出了各种编码器和解码器。 最后,采用多关系图卷积网络编码器和神经多关系解码器(NMR-RGCN)的NMCMDA获得了最佳的预测性能。 在三个实验数据集上,我们将所提出的方法与其他基线进行了比较。 实验结果表明,NMR-RGCN在Top-1准确率、Top-1查全率和Top-1 F1方面明显优于TDRC方法。 此外,我们还对两种高危疾病(乳腺癌和肺癌)进行了案例研究,并根据HMDD V3.2的所有已知数据对前10个miRNA疾病类别关联进行了预测和验证,进一步验证了该方法的有效性和可行性。
3.材料(material)
人类 MiRNA 疾病数据库 (HMDD) [26, 27] 是一个包含经过实验验证的人类 miRNA 与疾病关联的数据库。自 2007 年发布第一个版本以来,HMDD 一直是 miRNA-疾病关联研究的重要数据源。 HMDD 为 miRNA 与疾病之间的关联提供了多种类型的证据,这些证据在理解导致疾病的miRNA 失调的潜在机制方面发挥着重要作用。这些关联数据为数据驱动的MDA研究提供了重要的数据基础。此外,其他辅助信息,如 miRNA-miRNA 相似性和疾病-疾病相似性,可用于提高 MDA 预测性能。
在接下来的内容中,我们首先介绍HMDD中多个关联类别的详细信息。 然后讨论了从相应的数据源中构造miRNA-miRNA相似度和疾病-疾病相似度的方法。
3.1.miRNA与疾病的多种关联(Multiple categories of MiRNA–disease associations)
最近发布的HMDD V3.2提供了6个广义的关联类别(遗传学、表观遗传学、靶点、循环、组织和其他),涵盖了20种详细的证据代码。 经过预处理和去除重复,我们最终获得以下数据集:
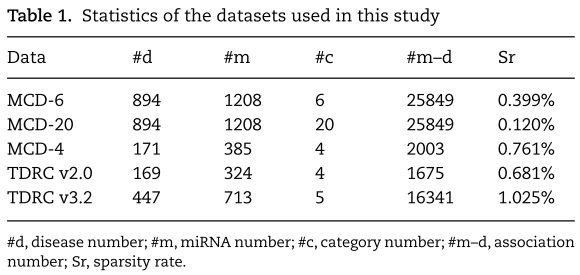
- MCD-6来自HMDD V3.2,包含上述6类,包括894种疾病与1208个miRNAs之间的25849个关联。
- MCD-20也是从HMDD V3.2中获得的,它包含了关于894种疾病和1208种miRNAs之间关联的20个详细的证据代码。
- 此外,我们还采用了以下数据集作为实验数据集来比较我们提出的方法的性能。
- MCD-4来自于2013年发布的HMDD V2.0[26]。 MCD-4包含了324个miRNAs与169种疾病之间的联系的四个类别(遗传学、表观遗传学、循环和靶点)。
- TDRC V2.0 和TDRC V3.2 由已发表的论文[35]发布和提供。 TDRC V2.0包含了169种疾病和324个miRNAs之间的四大类(遗传学、表观遗传学、循环和靶点)关联。 TDRC V3.2包含了447种疾病和713个miRNAs之间的五大类(遗传学、表观遗传学、循环、靶点和组织)关联。
这些数据集的统计数据见表1。
我们观察到HMDD中所有miRNA与疾病的关联可以分为不同的类别。 补充表1分别给出了MCD-6、MCD-4和TDRC V3.2中不同类别的 m-d 关联的统计。 另一方面,某些miRNA与疾病的关联可能在不同的类别中同时发生。 补充表2给出了在不同类别中同时出现的疾病-miRNA关联的统计。 我们可以看到,虽然许多miRNA与疾病的关联只属于一个类别,但有些仍然属于两个或两个以上的类别。 这进一步增加了准确预测的挑战。

3.2.MiRNA–miRNA similarity
在本研究中,利用miRNA的功能相似度得分和高斯互作谱核相似度得分来衡量miRNA的相似度。 miRNA i 和 j 之间的miRNA功能相似性定义如下:

其中![]() 是从MISIM2.0数据库(http://www.lirmed.com/misim/)下载的功能相似性分数。
是从MISIM2.0数据库(http://www.lirmed.com/misim/)下载的功能相似性分数。![]() 是高斯互作谱核相似度得分[6,13,36,37],用于补充MISIM中缺失的条目。 具体地说,
是高斯互作谱核相似度得分[6,13,36,37],用于补充MISIM中缺失的条目。 具体地说,![]() 计算如下:
计算如下:
![]()
其中![]() 表示相邻矩阵T中的 i 行,
表示相邻矩阵T中的 i 行,![]() 是核带宽参数,由下式计算:
是核带宽参数,由下式计算:
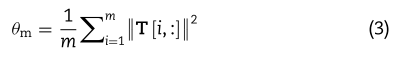
其中m是miRNAs的数目,即T的行数。
对于![]() ,miRNA功能相似度矩阵表示为
,miRNA功能相似度矩阵表示为![]() ,根据
,根据![]() 创建。
创建。
3.3.疾病-疾病相似性(Disease–disease similarity)
基于疾病层次有向无环图(DAG),计算疾病-疾病语义相似度得分(https://www.nlm.nih.gov/mesh/)。 首先,令 i 是一种疾病。 dag(i)表示疾病DAG中的节点集,包括节点 i 及其祖先节点。 然后,疾病 t 对疾病 i 的第一语义贡献由![]() 表示,并且可以使用下面的方程式求出[38],
表示,并且可以使用下面的方程式求出[38],
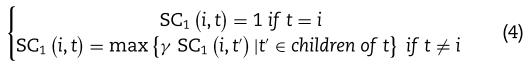
其中γ是语义贡献衰减因子,表明随着疾病 t 与其祖先疾病之间距离的增加,它们对疾病 d 语义值的贡献逐渐减小。 根据文献[37]将γ设为0.5。
基于式(4)中语义贡献的定义,建立了以![]() 表示的不同疾病之间的第一语义相似度评分。 设
表示的不同疾病之间的第一语义相似度评分。 设![]() 是两种不同的疾病。
是两种不同的疾病。 ![]() 定义如下:
定义如下:
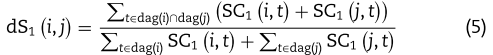
直观地说,如果 DAG 的大部分由 i 和 j 共享,![]() 就更高。
就更高。
然而,![]() 忽略了不同疾病贡献的重要性。 假设
忽略了不同疾病贡献的重要性。 假设![]() ,如果疾病
,如果疾病![]() 只出现在
只出现在![]() 中,而
中,而![]() 同时出现在
同时出现在![]() 和其他疾病的DAG中,则
和其他疾病的DAG中,则![]() 对
对![]() 的语义贡献可能高于
的语义贡献可能高于![]() 。 因此,第二语义贡献分数
。 因此,第二语义贡献分数![]() 下所示:
下所示:

基于![]() ,给出了两种疾病之间的第二语义相似度评分
,给出了两种疾病之间的第二语义相似度评分![]() ,如下[38]:
,如下[38]:
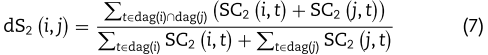
由于使用![]() 和
和![]() 计算的疾病相似性度量均来自 MeSH 数据库,因此它仅提供疾病语义相似性矩阵中的一部分条目。因此,采用高斯互作谱核相似度来补充剩余的疾病相似度条目。
计算的疾病相似性度量均来自 MeSH 数据库,因此它仅提供疾病语义相似性矩阵中的一部分条目。因此,采用高斯互作谱核相似度来补充剩余的疾病相似度条目。
设![]() 为利用已知的HMDD V2.0 miRNA与疾病关联数据构造的邻接矩阵。
为利用已知的HMDD V2.0 miRNA与疾病关联数据构造的邻接矩阵。 ![]() 是表示疾病
是表示疾病![]() 的
的![]() 二元向量。 然后,将疾病
二元向量。 然后,将疾病![]() 和疾病
和疾病![]() 之间的高斯互作谱核相似度定义为:
之间的高斯互作谱核相似度定义为:
![]()
其中![]() 是使用以下公式计算的内核带宽参数:
是使用以下公式计算的内核带宽参数:

其中n是疾病的数目,即T的列数。
在![]() 条件下,疾病语义相似度矩阵用
条件下,疾病语义相似度矩阵用![]() 表示,构造如下
表示,构造如下

4.方法(Method)
在这一节中,我们首先将多类别miRNA与疾病的关联预测表述为一个张量补全问题。 然后,提出了一种基于端到端学习的预测模型来解决该问题。
4.1.问题表述(Problem formulation)
多类别关联可以被组织成一个二元三维张量来表示![]() ,其中
,其中![]() 和
和![]() 分别表示miRNAs集合的大小、疾病集合的大小和关联类别的大小。
分别表示miRNAs集合的大小、疾病集合的大小和关联类别的大小。![]() 的
的![]() 切片是关于
切片是关于![]() -类别已知miRNA-疾病关联的含有0-1项的邻接矩阵
-类别已知miRNA-疾病关联的含有0-1项的邻接矩阵![]() ,其中
,其中![]() ,表示在
,表示在![]() -证据类别中一个miRNA
-证据类别中一个miRNA![]() 与一个疾病
与一个疾病![]() 关联。 如果miRNA i 与疾病 j 之间的联系未知或未观察到,
关联。 如果miRNA i 与疾病 j 之间的联系未知或未观察到,![]() =0。
=0。
多类别 miRNA 疾病关联预测(以下简称 MCMDA)问题可以用 m 个miRNA 和 n 种疾病来考虑,部分观察到的 m × n × R 三维关联张量 ![]() 其中每个条目
其中每个条目 ![]() 表示 miRNA i 在 r 类证据中与疾病 j 相关。如果 miRNA i 和疾病 j 之间的关联在 r 类证据中未知或未观察到,则
表示 miRNA i 在 r 类证据中与疾病 j 相关。如果 miRNA i 和疾病 j 之间的关联在 r 类证据中未知或未观察到,则![]() 。然后,MCMDA的目标是寻找一个近似张量
。然后,MCMDA的目标是寻找一个近似张量![]() ,如下:
,如下:

其中![]() 是张量Fresenius范数,定义为
是张量Fresenius范数,定义为![]()
![]() 。
。![]() 是一个索引集,表示观测值的索引。
是一个索引集,表示观测值的索引。 ![]() 是
是![]() 在集合
在集合![]() 上的投影。 对等式(11)的直观解释是:对于MCMDA,我们期望找到一个张量
上的投影。 对等式(11)的直观解释是:对于MCMDA,我们期望找到一个张量![]() ,它服从于由观察值(也就是实验验证的关联)所给出的同等约束。
,它服从于由观察值(也就是实验验证的关联)所给出的同等约束。
我们注意到等式(11)的目标只考虑了实验验证的关联。 从表1中,我们知道![]() 的sparsity rate非常低,这意味着只有少数实验验证的 miRNA-疾病 条目可以被观察到。
的sparsity rate非常低,这意味着只有少数实验验证的 miRNA-疾病 条目可以被观察到。 ![]() 中有许多未知或未被观察到的条目。需要指出的是,'未知‘并不意味着’没有相互作用‘。 “未知”包括两种情况,一种是“没有相互作用”,另一种是“相互作用但我们还不知道”。 在优化目标中忽略这些条目可能会产生退化的结果。 为了弥补这一缺陷,在目标函数中应该考虑观察到的条目和未知(或未观察到的)条目之间的平衡权衡。 此外,还可以利用其他有关疾病和miRNAs的辅助信息来进一步提高预测性能。 此外,还应建立参数化模型,以产生
中有许多未知或未被观察到的条目。需要指出的是,'未知‘并不意味着’没有相互作用‘。 “未知”包括两种情况,一种是“没有相互作用”,另一种是“相互作用但我们还不知道”。 在优化目标中忽略这些条目可能会产生退化的结果。 为了弥补这一缺陷,在目标函数中应该考虑观察到的条目和未知(或未观察到的)条目之间的平衡权衡。 此外,还可以利用其他有关疾病和miRNAs的辅助信息来进一步提高预测性能。 此外,还应建立参数化模型,以产生![]() 。 总之,MCMDA可以更准确地定义如下:
。 总之,MCMDA可以更准确地定义如下:
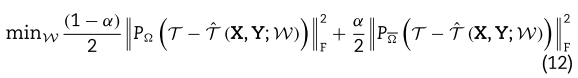
其中参数![]() 是一个偏差项,它适当地加权了观察项和未观察项。
是一个偏差项,它适当地加权了观察项和未观察项。 ![]() 表示
表示![]() 中未知或未观察项的子集集合。
中未知或未观察项的子集集合。![]() 是产生
是产生![]() 的可训练参数。 图1显示了MCMDA的一般流程图。
的可训练参数。 图1显示了MCMDA的一般流程图。
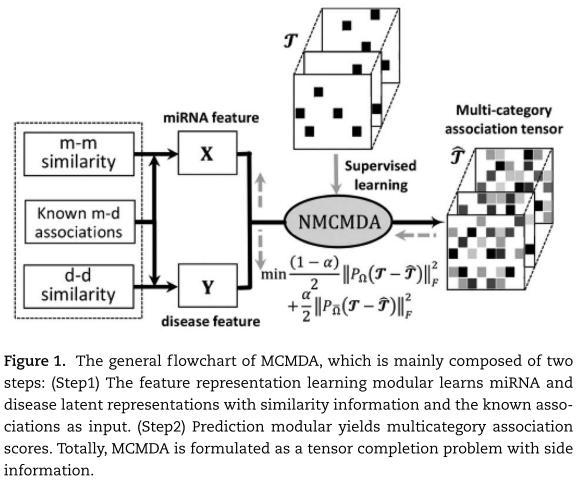
当我们求解方程(12)定义的MCMDA时,必须回答一些问题。 首先,miRNAs和疾病的适当特征表示是什么? 如何利用miRNA与miRNA的相似性、疾病与疾病的相似性以及已知的不同类别的miRNA与疾病的关联来获得特征矩阵X和Y? 二是如何捕捉潜在特征表示与不同关联类别之间的依赖关系,并建立设计良好的参数化模型以产生预测得分张量![]() ? 最后,能否有效地整合以上两个模块,构建从miRNA和疾病特征表示学习到多关联预测的端到端学习管道?
? 最后,能否有效地整合以上两个模块,构建从miRNA和疾病特征表示学习到多关联预测的端到端学习管道?
为了回答这些问题并解决MCMDA的问题,本文提出了一种基于端到端学习的预测模型,该模型直接作用于MIRNA疾病的多关系异构图。 一般来说,我们提出的模型有两个主要组成部分:(一)编码器利用图神经网络在miRNA-疾病多关系异构图上分别学习miRNA和疾病的潜在表征。 (二)解码器以学习到的潜在表征作为输入,产生miRNA-疾病关联分数。 在下面,我们讨论两个组件的细节。
4.2.miRNA与疾病潜在特征的编码器(Encoders for latent feature of MiRNA and disease)
本部分提出了两种用于获取miRNAs和疾病潜在特征的编码器:第一种是GCN-编码器,它分别在miRNA-miRNA相似网络和疾病-疾病相似网络上使用图卷积网络将miRNAs和疾病的节点映射到潜在特征。 第二种是RGCN-编码器,它利用相似度网络和已知的miRNA与疾病关联的不同类别来产生潜在特征。
4.2.1.GCN-编码器
让我们从讨论GCN编码器的细节开始。 miRNA与疾病关联预测的基本假设是功能相似的miRNA更有可能与表型相似的疾病相关,反之亦然。 因此,miRNAs与疾病之间的相似性信息对MDA至关重要。 这里,利用图卷积网络(GCN)[39]在miRNA(或疾病)相似性网络上给出了 miRNA和疾病的潜在特征表示。
设![]() 和
和![]() 分别为miRNA功能相似网络和疾病语义相似网络。
分别为miRNA功能相似网络和疾病语义相似网络。 ![]() 表示
表示![]() 的邻接矩阵,
的邻接矩阵,![]() 表示
表示![]() 的邻接矩阵。
的邻接矩阵。![]() 和
和![]() 分别表示
分别表示![]() 上的节点集
上的节点集![]() 和
和![]() 上的节点集
上的节点集![]() 的大小。 设
的大小。 设![]() 和
和![]() 分别是图
分别是图![]() 和
和![]() 结点集上的初始特征。 GCN通过分层聚合节点 i 邻域的特征信息来学习节点 i 的特征。 接下来,我们介绍了
结点集上的初始特征。 GCN通过分层聚合节点 i 邻域的特征信息来学习节点 i 的特征。 接下来,我们介绍了![]() 上miRNAs的特征学习方法,
上miRNAs的特征学习方法,![]() 上diseases的特征学习方法也是类似的过程。
上diseases的特征学习方法也是类似的过程。
具体来说,GCN中节点 i 的单层特征聚合算子定义为:
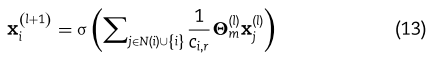
其中![]() 是
是![]() 层miRNA节点
层miRNA节点![]() 的隐藏表示。
的隐藏表示。![]() 表示具有 self-loop 的
表示具有 self-loop 的![]() 的相邻矩阵,其中
的相邻矩阵,其中![]() 为单位矩阵。
为单位矩阵。 ![]() 表示具有
表示具有![]() 的对角矩阵。
的对角矩阵。![]() 是具有
是具有![]() 输入通道和
输入通道和![]() 滤波器的滤波器参数矩阵。 σ(·)是非线性激活函数,如ReLU。
滤波器的滤波器参数矩阵。 σ(·)是非线性激活函数,如ReLU。 ![]() 是归一化常数。等价矩阵形式也可以这么写:
是归一化常数。等价矩阵形式也可以这么写:
![]()
使用![]() 表示
表示![]() 。 然后,多层特征聚集运算可以堆叠为一个 L 层GCNs,其表示如下:
。 然后,多层特征聚集运算可以堆叠为一个 L 层GCNs,其表示如下:
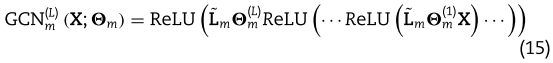
其中,![]() 是所有miRNAs的随机初始化的特征矩阵,而
是所有miRNAs的随机初始化的特征矩阵,而![]() 是总的可训练参数。 同样,对于疾病我们有
是总的可训练参数。 同样,对于疾病我们有
![]()
因此,考虑一个miRNA功能相似度网络和一个疾病语义相似度网络,从随机初始化的嵌入![]() 开始,GCN对特征进行逐层变换,最终输出
开始,GCN对特征进行逐层变换,最终输出![]() 和
和![]() 。 这些学习到的特征将作为下游多类别关联预测模型的输入。
。 这些学习到的特征将作为下游多类别关联预测模型的输入。
注意,GCN-编码器在产生潜在特征时忽略了miRNA和疾病之间已知的多种关联。 自然,通过考虑这些关联,潜在特征的质量可能会进一步提高。 事实上,miRNA、疾病相似网络以及已知的miRNA-疾病关联共同构成了异构的多关系网络。 因此,我们可以利用关系图卷积网络[40]来产生潜在的特征表示。
4.2.2.RGCN-encoder
RGCN-编码器也是一种基于图卷积网络的编码器,它通过利用miRNA和疾病的相似性网络和已知的不同种类的miRNA和疾病的关联来产生潜在的特征。 具体来说,RGCN编码器中用于miRNA i 的单层特征聚集算子定义为:

其中![]() 是miRNA i 的当前层隐藏表示,
是miRNA i 的当前层隐藏表示,![]() 是节点 i 的 r-类别邻居,
是节点 i 的 r-类别邻居,![]() 是一个归一化常数,
是一个归一化常数,![]() 是 r 类别关联关系的滤波参数矩阵,该关联关系由 i 的所有 r 类别关联邻居共享。
是 r 类别关联关系的滤波参数矩阵,该关联关系由 i 的所有 r 类别关联邻居共享。![]() 是每个节点本身的滤波器参数矩阵。像这样,我们还有一个针对疾病 i 的单层特征聚集算子,如下所示。
是每个节点本身的滤波器参数矩阵。像这样,我们还有一个针对疾病 i 的单层特征聚集算子,如下所示。
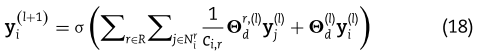
与GCN-编码器类似,我们可以将多层RGCN特征聚合操作叠加形成一个RGCN编码器。 miRNA和疾病的RGCN编码器分别用![]() 和
和![]() 表示。
表示。
与GCN-encoder的思想不同,RGCN-encoder充分考虑了miRNA与疾病之间不同类型的关系,如miRNA-miRNA相似性、疾病-疾病相似性、多类别miRNA-疾病关联等,使用特定关系的过滤器参数矩阵将特征转化为不同类型的特征空间,最终聚合不同类型的特征。我们将通过实验结果证明这会提高表示学习的质量。
4.3.译码器评分多类别miRNA与疾病关联
在我们提出的预测模型中,r 类别的关联预测分数由解码器获得,解码器是参数化得分函数![]() ,其中 X,Y分别是编码的miRNA和疾病特征。在本文中,我们介绍了以下三种不同的译码器。
,其中 X,Y分别是编码的miRNA和疾病特征。在本文中,我们介绍了以下三种不同的译码器。
DisMult 译码器(![]() )采用 DistMult 因式分解[41]作为评分函数,它在标准多类别链接预测问题上表现良好。在
)采用 DistMult 因式分解[41]作为评分函数,它在标准多类别链接预测问题上表现良好。在![]() 中,每个类别 r 与一个对角矩阵
中,每个类别 r 与一个对角矩阵![]() 相关联,r 类别下的miRNA-疾病对(x,r,y)被打分为
相关联,r 类别下的miRNA-疾病对(x,r,y)被打分为
![]()
我们观察到 DistMult 解码器中的对角矩阵![]() 仅捕捉特定 r 类别下的miRNAs与疾病之间的相互作用。 然而,如补充表2所示,他们可能存在不同类型的关联。 为此,我们对
仅捕捉特定 r 类别下的miRNAs与疾病之间的相互作用。 然而,如补充表2所示,他们可能存在不同类型的关联。 为此,我们对![]() 进行了扩展,将一个可训练参数矩阵 G 引入到
进行了扩展,将一个可训练参数矩阵 G 引入到![]() 中,并提出了下面的线性多关系译码器(Linear Multi-Relational decoder)。
中,并提出了下面的线性多关系译码器(Linear Multi-Relational decoder)。
LMR 译码器(由![]() 表示)定义如下。
表示)定义如下。
![]()
其中![]() 是一个参数矩阵,它描述了不同类别疾病和miRNAs的潜在特征的全局相互作用。
是一个参数矩阵,它描述了不同类别疾病和miRNAs的潜在特征的全局相互作用。 ![]() 仍然是一个可训练的对角矩阵,它捕捉了潜在表示中每个维度对 r 类别关联的重要性。
仍然是一个可训练的对角矩阵,它捕捉了潜在表示中每个维度对 r 类别关联的重要性。
![]() 和
和![]() 都是双线性译码器。 受NIMCGCN[25]的启发,我们提出了一种新的神经多类别关联评分模型来捕捉miRNAs与疾病的潜在特征之间更深层次的非线性相互作用。
都是双线性译码器。 受NIMCGCN[25]的启发,我们提出了一种新的神经多类别关联评分模型来捕捉miRNAs与疾病的潜在特征之间更深层次的非线性相互作用。
NMR-解码器(用![]() 表示)表示神经多关系解码器(Neural Multi-Relational decoder)。 下面我们将以miRNA为例来说明
表示)表示神经多关系解码器(Neural Multi-Relational decoder)。 下面我们将以miRNA为例来说明![]() 的思想,同样的思想也可以应用于疾病。
的思想,同样的思想也可以应用于疾病。
以GCN-output(或RGCN-output)特征![]() 作为输入,建立了一个K层前馈神经网络
作为输入,建立了一个K层前馈神经网络![]() 来进一步变换每一类 r 的miRNA的特征。 具体来说,
来进一步变换每一类 r 的miRNA的特征。 具体来说,
![]()
在![]() 定义了一种
定义了一种![]() 的非线性变换为
的非线性变换为![]() ,其中
,其中![]() 是
是 ![]() 层的特征矩阵,
层的特征矩阵,![]() 是转换参数矩阵,
是转换参数矩阵,![]() 是偏置向量。σ(·)是非线性激活函数。我们用
是偏置向量。σ(·)是非线性激活函数。我们用![]()
![]() 表示所有可训练参数,其中
表示所有可训练参数,其中![]() 是
是![]() 涉及到的参数。
涉及到的参数。
然而,上述特定类别的神经网络无法捕捉跨不同类别的潜在特征之间的相互作用。 为了解决这个问题,我们在特定分类神经网络之后建立了一个全局H层前向神经网络![]() 来进一步捕捉所有可能的不同类别的miRNA潜在特征的相互作用。 具体来说,
来进一步捕捉所有可能的不同类别的miRNA潜在特征的相互作用。 具体来说,
![]()
其中![]() 是第 r 个分类的特定神经网络输出的特征矩阵。
是第 r 个分类的特定神经网络输出的特征矩阵。![]() 和
和![]()
![]() 是不同类别的输入特征共享的全局可训练参数矩阵和偏差向量。
是不同类别的输入特征共享的全局可训练参数矩阵和偏差向量。
在得到所有miRNAs的![]() 和所有疾病的
和所有疾病的![]() 后,r 类关联预测分数矩阵
后,r 类关联预测分数矩阵![]() 的是
的是![]() 和
和![]() 的点积,即
的点积,即![]() 。
。
![]()
这样,我们就可以将上述各分量集成到一个统一的预测模型中,并用下面的点积计算出![]() 。
。
![]()
4.4.端到端学习模型(End-to-end learning models)
上一小节中描述的各种编码器和解码器可以组合成特定的MCMDA预测模型。 例如,当使用RGCN作为编码器,NMR作为解码器时,我们得到了NMR-RGCN。 同样,我们也有NMR-GCN、LMR-RGCN等。 接下来,我们引入了一个通用的损失函数,它可以作为不同编码器-解码器组合的损失函数。
现在,我们给出方程(12)中定义的MCMDA损失函数的细节。 具体而言,观察到的关联张量![]() 可以被描述为一组不同类别的关联矩阵,即
可以被描述为一组不同类别的关联矩阵,即![]() ,其中
,其中![]() 是实验验证的 r-类别miRNA-疾病关联矩阵。 对于r-类别,
是实验验证的 r-类别miRNA-疾病关联矩阵。 对于r-类别,![]() 和
和![]() 用于表示已知关联矩阵
用于表示已知关联矩阵![]() 中已观察到的和未观察到的或未知的 miRNA 疾病条目的集合。 观察值
中已观察到的和未观察到的或未知的 miRNA 疾病条目的集合。 观察值![]() 仅由正关联组成,即如果
仅由正关联组成,即如果![]() 。
。 ![]() 是未知项或未观测项的集合,如果
是未知项或未观测项的集合,如果![]() 。 利用这些符号,公式(12)中定义的MCMDA可以重新表述如下
。 利用这些符号,公式(12)中定义的MCMDA可以重新表述如下

其中![]() 由等式(24)计算,
由等式(24)计算,![]() 表示包含编码器和解码器参数的所有可训练参数。
表示包含编码器和解码器参数的所有可训练参数。
值得一提的是,编码器和解码器被集成到一个统一的端到端神经网络学习框架中。具体而言,GNN 编码器首先被利用于分别通过 miRNA-疾病异质信息网络学习 miRNA 和疾病特征。然后,解码器接收学习到的潜在特征进行进一步的变换。 通过变换后的特征点积得到最终的预测分数。 编码器和解码器中涉及的所有参数![]() 通过带有自适应矩估计的梯度下降同时优化[42]。 图2演示了NMR-RGCN的流程图。
通过带有自适应矩估计的梯度下降同时优化[42]。 图2演示了NMR-RGCN的流程图。

5.结果和讨论(Results and discussion)
5.1.实验设置(Experimental setup)
实验代码是基于开源机器学习框架PyTorch(PyTorch)实现的。 基于开源的深度学习图库(Deep Graph Library)实现了图神经网络编码器。 所有实验都是在Windows10操作系统上, 带有Intel W-2145 8核、3.7GHz CPU和64G内存的Dell Precision T5820工作站进行的。
在本研究中,设置了以下两个评价设置。
(1)![]() :我们将所有实验证实的miRNA-疾病-类别的三联体(作为阳性样本)随机分成10个子集。 在每次折叠中,一个子集和随机抽样的未知三元组(作为阴性样本)的一个相等大小的集合作为测试集,其余的子集和随机抽样的未知样本的一个相等大小的集合作为训练集。 请注意,我们非常小心地确保训练集和测试集不相互包含。 采用查准率-查全率(AUPR)曲线下的面积和接收机工作特性(AUC)曲线下的面积来评价各种预测方法的预测性能。
:我们将所有实验证实的miRNA-疾病-类别的三联体(作为阳性样本)随机分成10个子集。 在每次折叠中,一个子集和随机抽样的未知三元组(作为阴性样本)的一个相等大小的集合作为测试集,其余的子集和随机抽样的未知样本的一个相等大小的集合作为训练集。 请注意,我们非常小心地确保训练集和测试集不相互包含。 采用查准率-查全率(AUPR)曲线下的面积和接收机工作特性(AUC)曲线下的面积来评价各种预测方法的预测性能。
(2)![]() :在我们的建模问题中,每个miRNA-疾病对通过零个、一个或多个关联类型联系起来。在这种情况下,我们随机地将所有与至少一种类型的关联相关的miRNA-疾病对分成十个子集。 在每个折叠中,依次将一个子集作为测试集,其余子集作为训练集。 根据预测得分,对测试集中的每个miRNA-疾病对在所有关联类别下进行排序。 然后将得分最高的类别作为模型对测试样本的预测结果,计算precision(Top-1)、recall(Top-1)、F1-score(Top-1)。
:在我们的建模问题中,每个miRNA-疾病对通过零个、一个或多个关联类型联系起来。在这种情况下,我们随机地将所有与至少一种类型的关联相关的miRNA-疾病对分成十个子集。 在每个折叠中,依次将一个子集作为测试集,其余子集作为训练集。 根据预测得分,对测试集中的每个miRNA-疾病对在所有关联类别下进行排序。 然后将得分最高的类别作为模型对测试样本的预测结果,计算precision(Top-1)、recall(Top-1)、F1-score(Top-1)。
相对于二元miRNA-疾病关联预测,在miRNA-疾病多关联预测问题中,我们更注重关联类别的预测,而这正是![]() 的测试目标,因此我们将
的测试目标,因此我们将![]() 作为主要的实验设置。
作为主要的实验设置。
5.2.各种编码器-解码器组合的性能分析(Performance analysis of various encoder–decoder combinations)
我们提出的模型完全基于编码器和译码器两个组成部分,并提出了不同的编码器来获得输入数据的特征表示。根据输入的编码特征,提出了多个解码器来产生多个类别的关联分数。
在本小节中,我们在 MCD-6 和 MCD-20 数据集上进行了大量的实验,系统地比较了NMR-RGCN(NMR作为解码器,RGCN作为编码器,其他符号的含义相似)、DistMult-RGCN、LMR-RGCN、NMR-GCN的性能。比较结果见表2和表3。

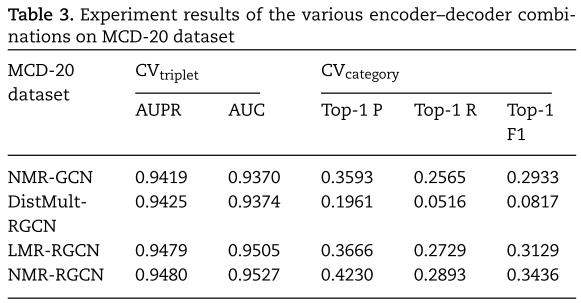
表2和表3中的实证结果显示了不同编码器和解码器对预测性能的影响。 具体而言,RGCN优于GCN,因为它利用了更多的链接信息来获得潜在的表示。此外,当使用 RGCN 作为编码器时,LMR 优于 DistMult,因为 LMR 学会捕获不同类别miRNA 和疾病的潜在特征的全局相互作用。此外,NMR 优于 LMR,因为 NMR 将 LMR 扩展成非线性神经网络框架。
由于NMR-RGCN的性能优于其他编码器-解码器组合,特别是在![]() 下,本文将NMR-RGCN作为主要的预测模型,讨论了不同参数对NMR-RGCN性能的影响,并将NMR-RGCN与其他基线进行了比较。
下,本文将NMR-RGCN作为主要的预测模型,讨论了不同参数对NMR-RGCN性能的影响,并将NMR-RGCN与其他基线进行了比较。
5.3.参数分析
以下参数将显著影响NMR-RGCN的性能:(i) α :方程(25)定义的损失函数中的偏差项;(ii) L :RGCN编码器的层数;(iii) K :由式(21)定义的特定类别神经网络的层数,(iv) H :由式(22)定义的遍及所有可能的不同类别的全局神经网络的层数。我们在MCD-6数据集上进行了实验,使用Top-1 precision、Top-1 Recall 和 Top-1 F1度量,以评估这些参数对NMR-RGCN性能的影响。
首先,引入有偏项 α 来适当地权衡观察项和未观察项。当 α = 0 时仅用正样本优化损失函数,当 α = 1 时仅用未观测样本优化损失函数。在本实验中,我们选定L=3, K=3, H=2。图3显示了不同 α 对NMR-RGCN预测性能的影响。 当 α = 0.2 时的性能优于当 α 设置为其他值时的性能。

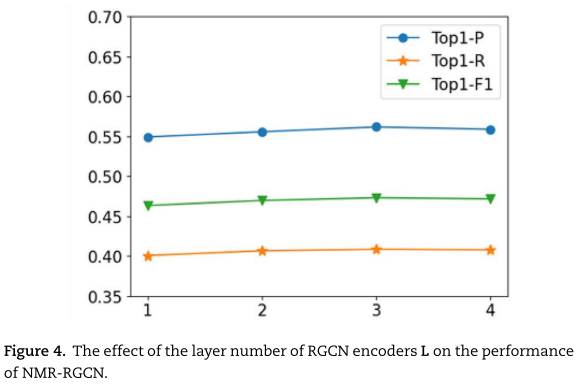
其次,我们分析了 L 对预测性能的影响。在本实验中,我们确定 α=0.2,K=3,H=2。 比较结果如图4所示。 从结果中,我们看到 L=3 的NMR-RGCN提供了最好的性能。 此外,我们还注意到,随着 L 的增加(L>3),由于较高的图卷积层对编码嵌入的过平滑,RGCN编码器的性能略有下降。
最后,如分段解码器评分多分类miRNA与疾病关联所描述的,神经多关系解码器由局部特定类别神经网络和含所有类别的全局神经网络组成。 在实验中,我们确定α=0.2,L=3。 我们测试以下情况:
(1)局部线性译码器+全局线性译码器。 在这种情况下,我们只考虑线性的局部解码器和全局解码器,即 K=1,H=1 并去掉非线性激活函数。 结果表明,线性译码器的性能远差于神经译码器。
(2)局部神经译码器。 我们去掉全局译码器,只考虑局部神经译码器。 从表4的实验结果中,我们发现当我们移除全局译码器时,模型的预测性能下降。 结果表明,全局解码器对于提高NMR-RGCN的预测性能是必要的。
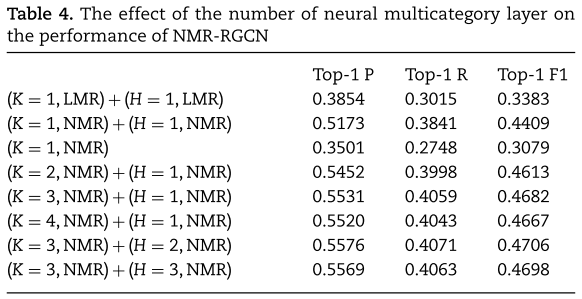
(3)局部神经解码器+全局神经解码器。 我们测试了局部神经解码器和全局神经解码器的几种组合的性能。 从表4的结果中,我们发现预测性能随着局部神经解码器层数的增加而提高。 但当K>3时,预测性能略有下降。 同样,对于全局神经投影,当H≤2时,预测性能随着层数的增加而提高。 注意,K=3,H=2的NMR解码器达到最佳性能。
5.4.与现有工作的比较(Comparisons with existing work)
在前一小节评价实验结果的基础上,我们用α=0.2,L=3,K=3和H=2作为NMR-RGCN的实验设置,用α=0.2,L=1,K=3和H=2作为NMR-GCN的比较实验设置。 自适应矩估计的学习速率为9e-4,调节系数为1e-8。
除了三种方法[33-35]外,我们还没有发现其他方法来预测多种类型的miRNA与疾病的联系。 在此,我们将NMR-RGCN(我们的方法)与NLPMMDA[34],TDRC[35]两种方法进行了比较。 NLPMMDA通过标签传播整合了miRNA相似性和疾病相似性,预测了多种关系的miRNA与疾病的关联。 在NLPMMDA中有两个超参数,我们根据原始文献分别设置![]() ,以获得最佳性能。TDRC引入了以miRNA功能相似度和疾病语义相似度为关系约束的张量分解,解决了多种类型的miRNA与疾病关联预测问题。 TDRC中有四个超参数,我们根据原始文献分别设置
,以获得最佳性能。TDRC引入了以miRNA功能相似度和疾病语义相似度为关系约束的张量分解,解决了多种类型的miRNA与疾病关联预测问题。 TDRC中有四个超参数,我们根据原始文献分别设置![]() ,以获得最佳性能。 所有对比实验分别在TDRC V3.2、TDRC V2.0 和 MCD-6 上进行。
,以获得最佳性能。 所有对比实验分别在TDRC V3.2、TDRC V2.0 和 MCD-6 上进行。
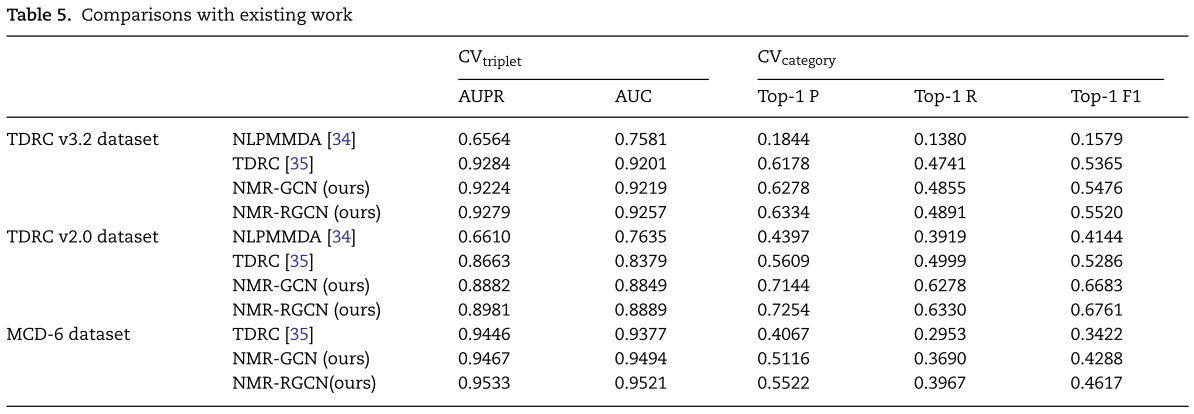
我们在表5中的总结了我们提出的模型和基线三个数据集上的实验比较结果。所有实验都是在上述两个实验设置下进行评估的。我们首先比较TDRC V2.0和TDRC V3.2数据集的结果。 我们发现NMR-RGCN、NMR-GCN和TDRC相对于NLPMMDA的改进尤为明显,这突出了考虑每种关联类型之间关系的重要性。然后,我们在MCD-6数据集上进一步比较了NMR-RGCN与TDRC和NMR-GCN。 我们观察到NMR-RGCN与TDRC相比,在Top-1精度方面提高了35.78%,在AUC方面提高了1.51%。这表明捕获多种类型的 miRNA 与疾病之间的非线性关系可以使我们获得更好的结果,因为 TDRC 本质上是一种多线性方法,可能不足以捕获 miRNA 与疾病特征之间的复杂和非线性相互作用。与NMR-GCN相比,NMR-RGCN的平均相对Top-1 precision提高了7.94%,这是因为该算法在对节点信息进行编码时还考虑了异构网络中其他来源的异构信息。
5.5.案例研究(Case studies)
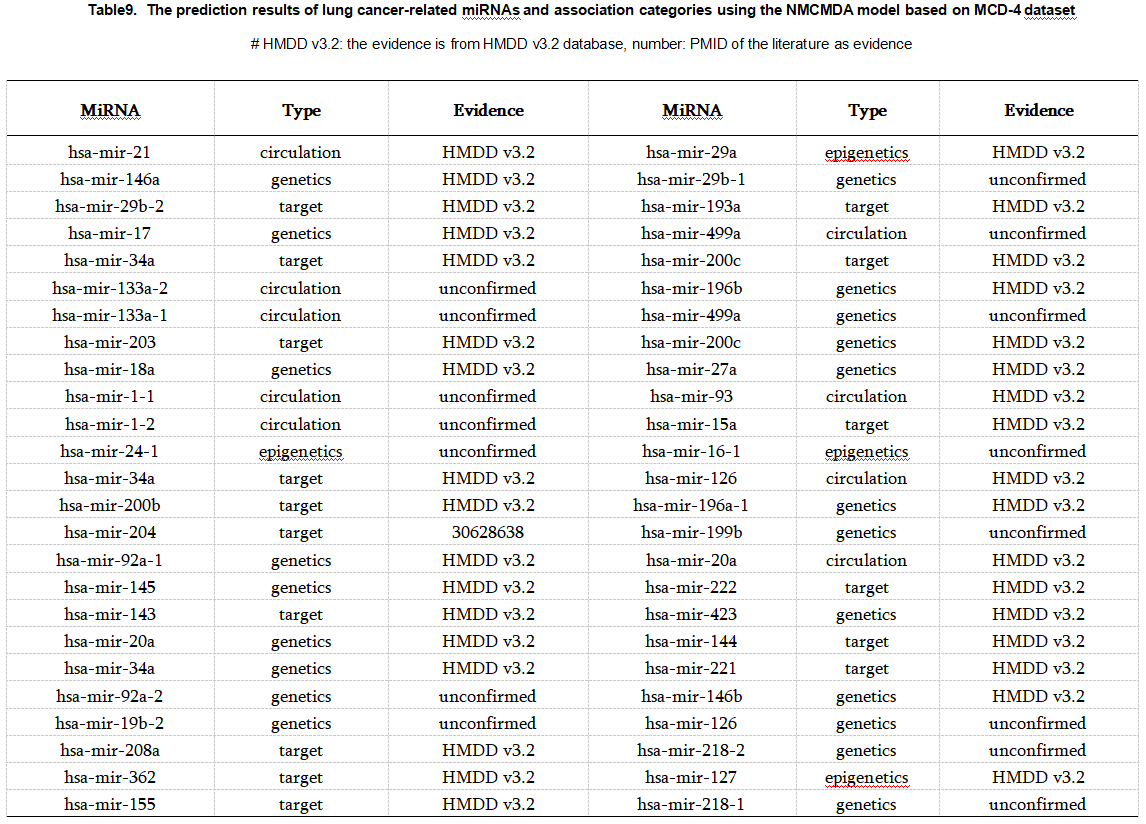
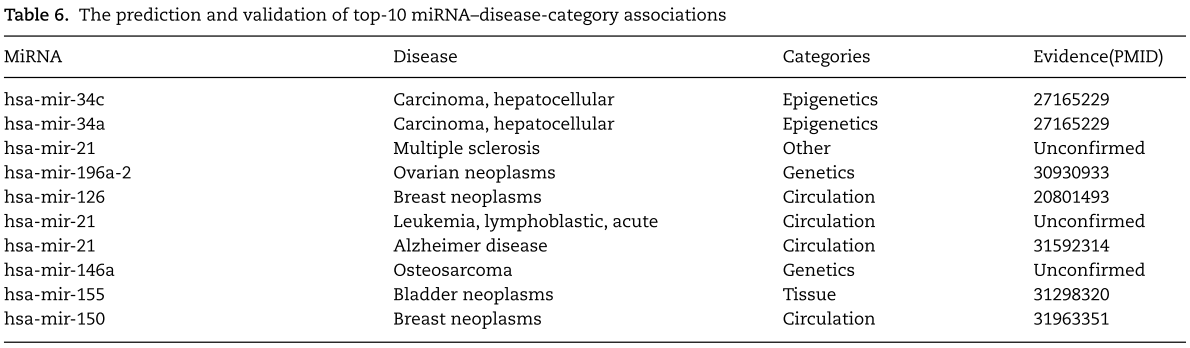
为了进一步评估我们提出的预测未观察到的 miRNA-疾病-类型 条目的模型的准确性,我们对两种广泛的人类疾病进行了案例研究,即乳腺癌和肺癌(补充表3和4)。 我们使用基于MCD-4数据集(所有已知的HMDD V2.0数据)的训练模型对特定疾病的候选miRNA类型对进行优先级排序,然后用HMDD V3.2数据集和最近的文献验证Top-50预测。 我们还基于MCD-6数据集(所有已知的HMDD V3.2数据)训练RGCN-NMR模型,然后使用它们的预测分数对所有未知的 miRNA-疾病-类型 条目进行优先级排序。 表6显示了前10个预测结果,根据最近的文献可以证实7个预测。 结果证明了我们提出的模型是有效的。

6.结论(Conclusion)
利用计算方法识别潜在的多类别miRNA与疾病的关联是重要的,因为它将提高我们对疾病发病机制的理解并指导治疗。在本研究中,我们开发了一种新的基于数据驱动的端到端学习的神经多类miRNA疾病关联预测方法NMCMDA来解决MCMDA问题。 NMCMDA在多类别miRNA与疾病关联的预测中表现出优异的性能,在Top-1 precision、Top-1 recall 和Top-1 F1方面显著优于TDRC[35]。 就我们的理解而言,NMCMDA相对于TDRC[35]的优势可能在于以下两个方面。 首先,NMCMDA是一个端到端的学习框架,编码器直接在miRNA疾病异构网络上操作,并利用GNN学习潜在的表征。解码器产生以学习到的潜在表征为输入的 miRNA-疾病关联分数。编码器和解码器中涉及的所有参数通过梯度下降同时优化。 而TDRC不是一种端到端的学习算法,不能保证潜在特征与预测目标直接相关。 第二,TDRC本质上是一种多线性方法,可能不足以捕捉miRNAs特征与疾病之间复杂和非线性的相互作用。 此外,我们还根据HMDD V3.2的所有已知数据,预测并验证了前10位miRNA疾病相关性,进一步验证了该方法的有效性和可行性。 为两种高风险人类疾病乳腺癌和肺癌提供了案例研究) ,我们还提供了基于 HMDD v3.2的所有已知数据的前10个 miRNA 疾病类别关联的预测和验证,这进一步验证了所提出的方法的有效性和可行性。
今后的研究有几个方向。 首先,关于miRNA和疾病相似度网络的结构信息显著影响学习到的特征表示,进而影响最终的预测结果。 其他生物医学信息来源,如miRNA与基因相互作用和疾病与基因相互作用等,可能与miRNA与疾病关联的建模有关,我们希望研究将它们集成到模型中的效用。 由于我们提出的NMCMDA是任何异构网络中多关系链路预测的通用方法,因此将其应用于其他领域和问题将是有趣的,例如,多关系药物-药物相互作用的识别[43]。
7.Supplementary data
The implemented code and experimental dataset are available online at https://github.com/ljatynu/NMCMDA/。