点击蓝字 关注我们
VeloPro: 东京大学齐藤裕组揭示蛋白质翻译速率与结构特征关联模式的分析流程

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.148
● 2023年11月19日,日本产业技术综合研究所人工智能研究中心与日本东京大学计算生物医学专业齐藤裕组在iMeta在线发表了题为 “VeloPro: A pipeline integrating Ribo-seq and AlphaFold deciphers association patterns between translation velocity and protein structure features” 的研究文章。
● 本研究开发了一种整合Ribo-seq和AlphaFold的分析流程,揭示了翻译速率与蛋白质结构特征之间的关联模式。
● 第一作者:边遍
● 通讯作者:齐藤裕([email protected])
● 合作作者:熊谷俊高
● 主要单位:日本东京大学计算生物医学专业、日本产业技术综合研究所人工智能研究中心、日本早稻田大学-产业技术综合研究所计算生物大数据创新开放实验室、日本北里大学数据科学专业
亮 点
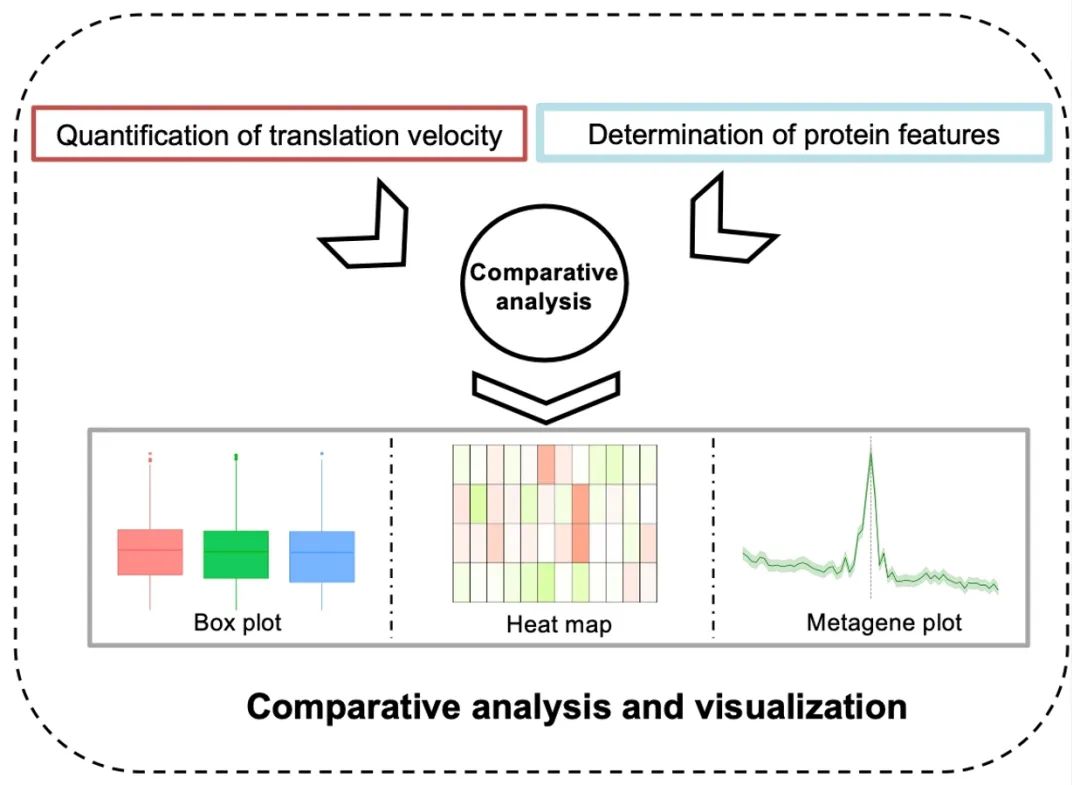
● VeloPro是一个易用的分析流程,使得能够在不同物种中进行翻译速率与许多蛋白质结构特征的关联分析;
● VeloPro通过关联分析阐明了翻译速率和蛋白质结构特征之间的关联模式在各种生物体是不同的,部分反映了它们的生物学分类关系,为精准RNA的设计及优化提供了新的思路。
摘 要
翻译速率,也称为核糖体滞留时间,在调节共同翻译蛋白质折叠和蛋白质功能完整性方面发挥着至关重要的作用。许多研究表明,翻译速率与某些蛋白质结构特征相关。然而,大多数研究集中在一些特定的生物体,如酵母和人类。在这里,我们开发了一个名为VeloPro的管道,通过整合公开可用的Ribo-seq数据和AlphaFold预测的三维蛋白质结构信息,来研究翻译速率与许多蛋白质结构特征之间的关联。这些特征包括蛋白质二级结构元素、带正电氨基酸、脯氨酸残基、相对可及表面积(rASA)、内在无序区(IDR)得分、局部接触顺序(反映蛋白质三维结构中氨基酸接触的局部性)以及密码子使用频率在12个不同分类群中如细菌、真菌、原生动物、线虫、植物、昆虫和哺乳动物。我们阐明了翻译速率与蛋白质结构特征之间的关联模式在各种生物体中存在差异,部分反映了它们的分类关系。我们发现,在酵母中先前报道的翻译速率与带正电氨基酸的关联方式在原核生物中不适用。我们还发现,大多数生物体中二级结构、rASA和局部绝对接触顺序的关联模式是广泛保守的。总的来说,我们的研究为理解跨分类群的共翻译折叠中的翻译动态提供了新的见解。我们的研究还可以在RNA设计中发挥作用,以微调共同翻译折叠,从而提高在不同寄主生物体中的重组蛋白表达。
视频解读
Bilibili:https://www.bilibili.com/video/BV1wc411q7K2/
Youtube:https://youtu.be/-BYE2AWdGjY
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
想要解读隐藏在翻译速率是蛋白质生物合成的关键过程,而翻译速率(也称为核糖体滞留时间)在调节蛋白质共同翻译折叠中发挥着至关重要的作用。蛋白质共翻译折叠过程始于新生肽链在核糖体出口通道内部出现的时候。翻译速率的变化可以通过在核糖体出口通道内部形成结构中间体来调节蛋白质的折叠。此外,共翻译折叠是通过蛋白质与核糖体出口通道的壶口之间的静电相互作用来促进的。许多研究表明,具有更复杂结构特性的较大蛋白质可能更依赖共同翻译折叠来避免错误折叠和聚集,这进一步说明了共同翻译折叠的至关重要性。核糖体通过将新生链穿过由同一蛋白质的较早合成的段创建的环,积极参与缠结蛋白质的折叠;因此,共同翻译折叠过程对于实现其多肽链深度缠结的蛋白质的天然结构可能是必要的。
随着深度测序技术的迅速发展,一种革命性的方法,核糖体测序(也称为Ribo-seq),已被用于在体内探测和定量在密码子分辨率水平上的局部翻译速率。由于Ribo-seq印记密度被认为与局部翻译速率呈负相关,Ribo-seq已成功用于了解细菌和真核生物中的局部翻译速率。
众所周知,翻译速率与RNA序列特征相关。许多开创性研究表明,Shine–Dalgarno(SD)类似序列强烈阻碍翻译,而一些研究认为SD类似序列motif对翻译延伸影响较小[18]。密码子使用偏好是影响翻译动力学的另一个重要因素。密码子使用偏好被认为与体内tRNA浓度相关,而稀有密码子会导致核糖体在翻译过程中停滞在mRNA上。此外,mRNA二级结构会影响翻译速率,因为mRNA的发夹结构可能会在翻译过程中阻碍核糖体的运动。最近,田岛等人发现翻译效率与mRNA序列特征之间的关系在九个微生物中存在差异,部分反映了它们的分类学差异。
尽管对RNA序列特征的研究日益增多,但很少有研究调查蛋白质特征与翻译速率之间的关联。周等人观察到在真菌Neurospora中编码α螺旋或β折叠的RNA区域更倾向于使用频繁的密码子,而编码无规则卷曲的RNA区域更倾向于使用稀有密码子。先前的研究还表明,在编码蛋白质无规则卷曲和结构化区域的RNA区域之间,翻译速率是不同的;在酵母和人类的mRNA序列中,编码无规则卷曲区域的RNA序列的翻译速率较慢,而编码α螺旋或β链区域的RNA序列的翻译速率较快,同时他们提出密码子使用偏好是解释这些差异的因素。此外,先前的研究表明,在酵母中,带正电氨基酸是翻译速率的主要抑制因素,并且在拟南芥叶绿体中也会导致核糖体停滞。另一项研究表明,在酵母中,一些短的新生肽序列,如PPP和LKK氨基酸序列,可能与核糖体在mRNA上停滞有关。然而,先前的研究主要集中在一些特定的生物体,主要是酵母和人类,引发了一个问题,即这些关联模式在不同的生物体中有多大程度的适用性。在更广泛的生物学分类学范围内揭示翻译速率与蛋白质特征之间的关联将有助于我们更好地理解可能影响共同翻译折叠的因素。
在本研究中,我们开发了VeloPro,这是一个集成公开可用的核糖体测序数据和AlphaFold预测的三维蛋白质结构信息的分析流程,用于研究12个不同分类群的生物体中翻译速率与蛋白质结构特征之间的关联模式,包括细菌(大肠杆菌和铜绿假单胞菌)、真菌(酿酒酵母和白念珠菌)、原生动物(布鲁氏锥虫)、线虫(秀丽隐杆线虫)、植物(拟南芥和玉米)、昆虫(果蝇)和哺乳动物(人类、大鼠和小鼠)。对于每个生物体,我们分析了比以前的研究更多的蛋白质特征,包括不同的蛋白质二级结构元素、脯氨酸残基、带正电氨基酸、相对可及表面积(rASA)、内在无序区(IDR)得分和局部接触顺序(local contact order)。我们发现翻译速率与蛋白质结构特征之间的关联模式在生物体间存在差异,部分反映了它们的生物学分类关系。我们发现在酵母中先前报道的翻译速率与带正电氨基酸的关联方式不适用于原核生物。我们还发现在大多数生物体中,蛋白质二级结构、rASA和局部绝对接触顺序(local absolute contact order)与翻译速率的关联模式是广泛保守的。
结 果
VeloPro的分析流程
在这项研究中,我们开发了一个名为VeloPro的流程,通过结和核糖体测序和AlphaFold预测的蛋白三维结构数据,然后调查了翻译速率与蛋白结构特征之间的关联模式。该分析流程在图1中有详细描述。
对于核糖体测序分析,先前的研究表明,Ribo-seq结果受到许多因素的影响,包括生长条件、细胞或组织的异质性、测序覆盖深度和误差、人为实验误差以及未知的生物体特异性因素。此外,先前的研究报告指出,一些生物信息学分析方法差异可能导致相互矛盾的结论。因此,在我们的分析中,我们采用了统一的生物信息学分析流程,用于所有生物体中的所有Ribo-seq数据分析和蛋白特征量化。此外,为了通过Ribo-seq数据分析量化翻译速率,我们采用了严格的偏移识别和校准方法(图1A)。
在我们的分析流程中,我们首先通过FastQC检查FASTQ文件的质量(支持信息:表S3),然后进行reads的比对。随后得到的BAM文件并被输入到校准过程中,以获得被认为与翻译速率呈反比的标准化印迹(图1A)。然后,利用AlphaFold对蛋白质预测的三维结构信息,计算蛋白结构特征,包括蛋白质二级结构元素、rASA、IDR分数和局部接触顺序(图1B)。接下来,我们对翻译速率和蛋白结构特征(包括rASA、IDR分数和局部接触顺序)进行了比较分析,以及对脯氨酸残基和带正电氨基酸进行了元基因分析。
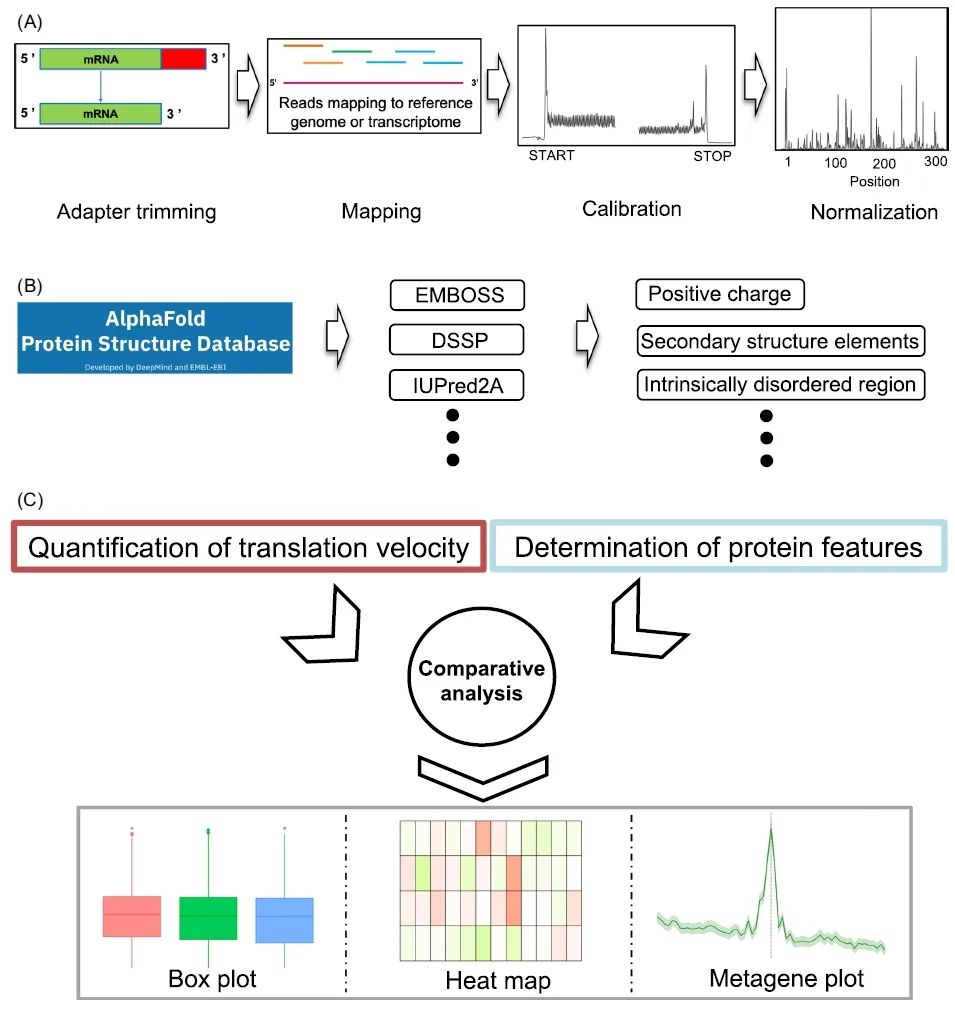
图1. VeloPro的工作流程
(A) 包括adapter修剪、比对、校准和归一化在内的翻译速率定量的流程。(B) 从AlphaFold获取蛋白质结构特征的流程。(C) 本研究中用于翻译速率和蛋白质结构特征之间的比较数据分析和可视化的示意图
通过核糖体测序分析在多个物种中对翻译速率进行量化
为了量化翻译速率以进行关联和元基因分析,我们分析了公开可用的核糖体测序的数据集(支持信息S2:表S2)。我们选择了包括细菌、原生动物、真菌、线虫、昆虫、植物和哺乳动物在内的12种生物,涵盖多个系统生物分类群。根据生物体的不同,有15到604百万个reads被唯一比对到参考序列上(支持信息S2:表S4和S5)。比对上reads的长度分布在每个生物体中都有所变化(支持信息S1:图S1,支持信息S2:表S4)。为了在每个生物体中校准印迹到核糖体P位点的位置,我们使用了MiMB_ribosome_profiling流程。对于每个数据集,我们提取了最丰富的reads长度 (支持信息S1:图S1)。然后,根据手册的指导,确定了每个读取长度的偏移量。确定的偏移值数据汇总在支持信息S2:表S4中。在校准后,在起始密码子和终止密码子周围生成了覆盖度图,所有生物体中CDS区域内均呈现三核苷酸周期性(支持信息S1:图S2)。这种三核苷酸周期性被认为是进行核糖体测序数据的密码子分辨率分析的良好数据质量的指标。我们使用这些覆盖度信息计算了标准化的核糖体印迹,用于量化每个密码子位置对应的翻译速率(详见补充材料和方法)。较少的核糖体印迹被认为表示较高的翻译速率,而较多的核糖体印迹则被认为表示较低的翻译速率或核糖体停滞。
在不同生物体中比较不同蛋白质二级结构元素之间的翻译速率
蛋白质在翻译过程中可能发生折叠,这被称为共翻译折叠过程。一些稀有密码子已与单个结构域或蛋白质中的二级结构的共翻译折叠中间体相关联,这表明在蛋白质合成过程中的暂停可能有助于共翻译折叠,从而促使其原生结构的形成。在这里,我们比较了不同蛋白质二级结构元素之间的翻译速率,以探究它们之间的关系。
在原核生物和大多数真核生物中,编码无规则卷曲区域的RNA序列的翻译速率显著慢于编码α-螺旋和β-折叠区域的序列(图2),这表明核糖体在编码非结构化区域的RNA序列中更容易停滞,而不是在编码结构化区域的RNA序列中。先前的研究报告中指出,编码非结构化区域的RNA序列中的停滞概率高于编码结构化区域的RNA序列,但该分析仅限于酵母和人类。我们揭示了这一模式在更广泛的生物体中是一致的。令人意外的是,在大鼠和布鲁氏锥虫中,编码无规则卷曲区域的RNA序列表现出比编码α-螺旋和β-折叠区域更快的翻译速率(图2E、K)。
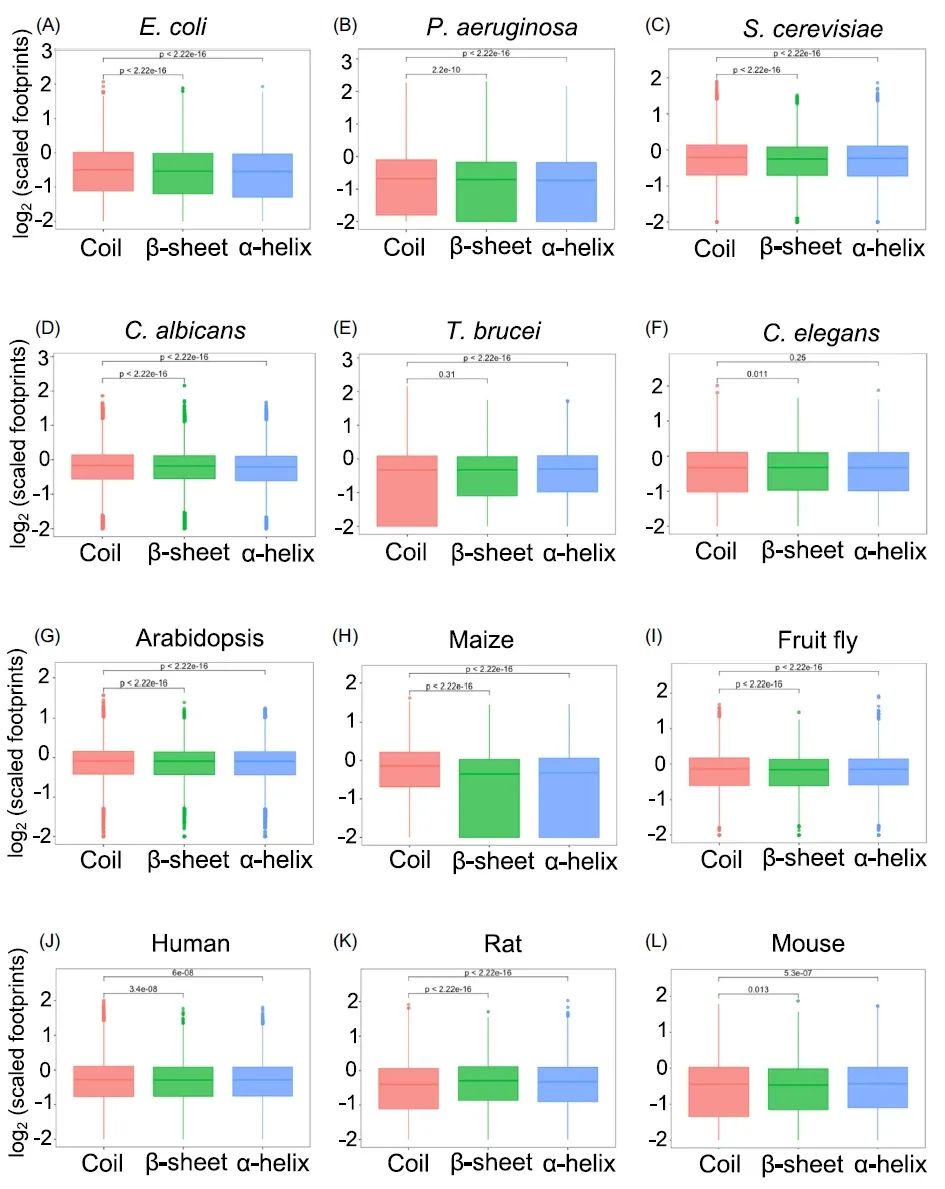
图2. 在12种生物中,比较了结构化区域(即α-螺旋和β-折叠)与无规则卷曲区域之间的标准化印迹
p值通过双侧Wilcoxon秩和检验计算得出
脯氨酸残基对翻译速率的影响是广泛保守的
先前的研究已经表明,脯氨酸作为肽键形成过程中的A位受体和肽基供体都表现较差,而且脯氨酸可能会减缓翻译过程中的肽键形成。许多先前的研究表明,氨基酸序列中的多脯氨酸片段在原核生物和真核生物中均可引起核糖体停滞。然而,很少有研究在单个氨基酸分辨率下分析脯氨酸残基对翻译速率的影响。
在我们对脯氨酸残基的元基因分析中,我们观察到在酵母和人类中脯氨酸残基周围存在明显的峰,表明我们的方法能够成功检测到由脯氨酸引起的停滞现象(支持信息S1:图S3C、J),这与先前的研究一致,即脯氨酸残基导致了人类和酵母中的核糖体停滞事件。此外,我们还观察到在许多真核生物中,如白念珠菌、拟南芥、玉米和果蝇,脯氨酸残基周围出现一个或多个峰(支持信息S1:图S3)。有趣的是,在原核生物中,如大肠杆菌和绿脓杆菌,我们并未观察到脯氨酸残基周围的峰值(支持信息S1:图S3A、B),尽管先前的研究声称脯氨酸-脯氨酸基序可能在细菌中引起核糖体停滞。
带正电氨基酸可能在原核生物和真核生物之间对翻译速率有不同的影响
在先前的研究中,已经表明新生肽链中的带正电氨基酸能够强烈抑制酵母中的翻译延伸速率。
我们在酵母中进行的元基因分析与先前的研究一致(支持信息S1:图S4C),显示在带正电氨基酸的位置周围存在明显的正峰。此外,我们在大多数真核生物中观察到了类似的在带正电氨基酸周围的正峰,如布鲁氏锥虫、白念珠菌、果蝇、拟南芥、玉米和人类(支持信息S1:图S4),这表明带正电氨基酸引起的核糖体停滞在真核生物中是广泛保守的。对于原核生物,先前并没有关于带正电氨基酸对翻译速率的影响的报道。我们发现,与真核生物中的情况相比,原核生物中的带正电氨基酸表现出了不同的方式,它们在元基因图中显示出负峰(支持信息S1:图S4A、B)。
在多种生物中,翻译速率和密码子使用频率之间的相关性分析
密码子使用已被证明影响局部翻译速率。稀有或非最优的密码子可能会减缓翻译速率,为蛋白质结构正确折叠提供足够的时间,从而精细调节共转译折叠过程。
我们在大肠杆菌、白念珠菌、果蝇、人类、大鼠和小鼠中进行的偏相关分析显示,在标准化印迹和密码子使用频率之间存在显著的负相关关系(图3;支持信息S2:表S6),这与先前的研究结果一致。然而,我们还观察到一个相反的结果,即在酵母、线虫和拟南芥中,密码子使用频率与标准化印迹之间存在显著的正相关关系(图3;支持信息S2:表S6)。这些结果表明,单独的密码子使用不能解释翻译速率的所有变异,暗示了可能存在的其他未知因素。为了探讨这些因素,我们进行了蛋白质特征和翻译速率之间的关联分析,如下面所述。
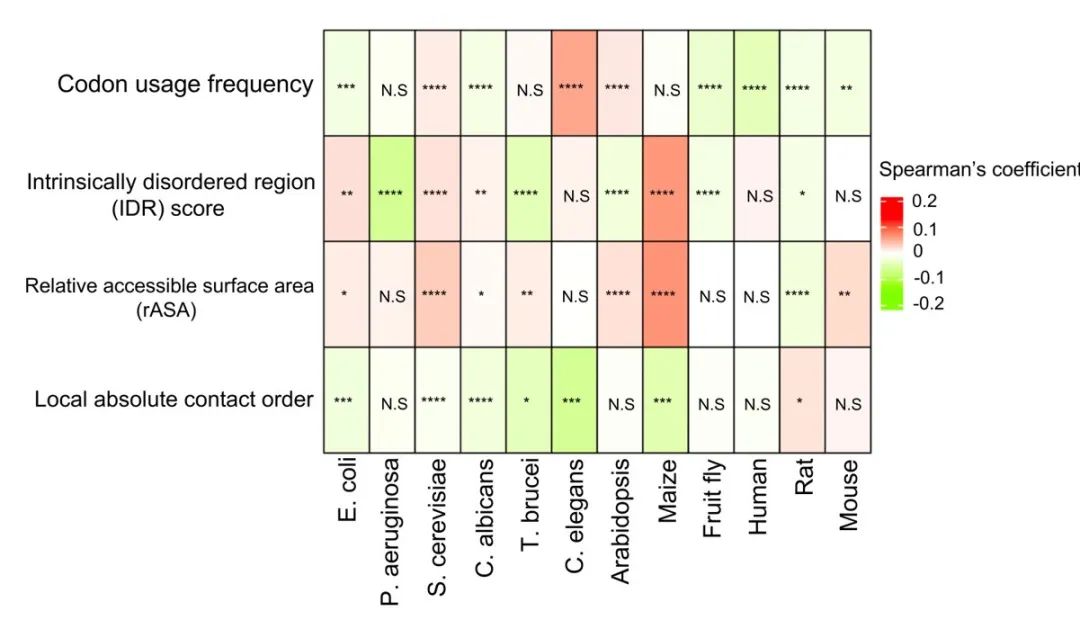
图3. 在12种生物体中,标准印迹与蛋白质结构特征之间的偏Spearman相关系数的热图
图中显示了每个物种 所有基因的偏相关系数均值。通过单样本t检验计算的多次检验的Bonferroni校正p值,用于检验均值与零的偏差:*p < 0.05,**p < 2.2e−5,***p < 2.2e−10,****p < 2.2e−15。N.S,不显著。均值的原始数值和Bonferroni校正的p值详见支持信息 S2: 表S6
在多种生物体中,翻译速率和IDR分数之间存在不同的关联模式
IDR(无序区域)被定义为不形成特定三维结构的序列片段。它们通常以富含极性或带电氨基酸为特征,缺乏足够的疏水氨基酸以实现协同折叠。IDR通过充当平台或支架,在介导与其他分子的相互作用中发挥着至关重要的作用。在先前的研究中,使用密码子适应指数(CAI)作为翻译延伸速率的预测,研究了IDR与预测的翻译速率之间的关系。他们观察到在大肠杆菌、酵母和秀丽线虫中,CAI与蛋白质无序趋势之间存在显著的负相关关系。然而,尚无研究是基于核糖体测序的。由于我们的分析显示了根据二级结构的不同而产生的翻译速率差异(图2),我们还探讨了IDR是否与翻译速率相关。
在我们的偏相关分析中,我们观察到在大肠杆菌和酵母中,标准化的核糖体印迹与IDR分数之间存在显著的正相关关系(图3;支持信息S2:表S6),这种模式与先前基于CAI的研究结果相似。然而,在细菌类群中,我们还在绿脓杆菌中观察到了与大肠杆菌中相反的关联模式。在真菌类群中,酵母和白念珠菌都表现出标准化的核糖体印迹与IDR分数之间的正相关关系。在植物类群中,我们观察到拟南芥(负相关)和玉米(正相关)之间存在相反的模式。
此外,我们还探讨了蛋白质不同区域(包括N-末端、中间和C-末端)之间的翻译速率和IDR得分之间的相关性(支持信息S1:图S5;支持信息S2:表S7)。在N-末端区域,相关性总体上较弱,但对于一些生物体,检测到了显著的正相关关系。在C-末端区域,酵母和白念珠菌表现出显著的正相关关系,而拟南芥、果蝇和小鼠等较高级真核生物则表现出显著的负相关关系。在中间区域的相关性模式与使用整个蛋白质区域进行的分析保持一致。
综上所述,IDR得分与翻译速率之间的关联在不同生物体中存在差异。这可能反映了它们在进化上的差异,然而我们无法做出更深入的解释。
rASA(相对可及表面积)和绝对接触顺序(absolute contact order)与翻译速率之间存在保守的关联模式
rASA(相对可及表面积)是对蛋白质3D结构中氨基酸残基与溶剂分子接触的度量。根据rASA的值,蛋白质的界面可以分为核心、边缘和支持端。较低的rASA表示蛋白质结构内的残基被埋藏(核心),而较高的rASA表示残基暴露于溶剂中(边缘)。
在我们的偏相关分析中,大多数生物体在标准化印迹和rASA之间呈现出显著的正相关关系(图3;支持信息S2:表S6),这表明编码核心区域的RNA序列相对于编码边缘区域的序列更倾向于较快地进行翻译。唯一的例外是大鼠,它显示出负相关,与其他生物体相反。
接触顺序是蛋白质3D结构中氨基酸接触局部性的度量。较大的接触顺序值表示氨基酸倾向于与序列中具有大间隔的远离的氨基酸发生接触。接触顺序已被用来比较拓扑差异,并显示与许多蛋白质性质相关,如蛋白质折叠速率和过渡态位置。在我们的偏相关分析中,我们发现在大肠杆菌中,标准化印迹与局部绝对接触顺序之间存在显著的负相关关系(图3;支持信息S2:表S6)。这与先前的研究结果一致,即在大肠杆菌中,具有较低接触顺序的蛋白亚结构与保守的稀有密码子相关联。此外,我们发现在大多数生物体中,除了大鼠外,标准化印迹与局部绝对接触顺序之间存在显著的负相关关系(图3;支持信息S2:表S6)。
讨 论
翻译速率在调节蛋白质丰度和通过共翻译折叠确保蛋白质的功能完整性方面发挥着重要作用。先前的报告表明,翻译速率沿mRNA序列不是均匀的,并试图找到决定这些变化的序列特征。然而,大多数先前的研究集中在mRNA序列特征与特定模式生物中翻译速率之间的关联。最近,Tajima等人发现了跨9个微生物的蛋白质表达的mRNA序列决定因素的多样性和共同点。然而,他们没有探讨相关的蛋白质结构特征。蛋白质3D结构预测的最新革命使我们能够在多种生物体中全面调查结构信息。在我们的研究中,通过结合Ribo-seq和AlphaFold预测的蛋白质3D结构数据,我们揭示了许多与翻译速率相关的蛋白质结构特征。通过分析来自各种进化类群的12个生物物种,我们还确定了在许多生物体中显示保守关联模式的特征,同时揭示了一些具有不同关联模式的特征。因此,我们的研究提供了与翻译速率相关的蛋白质特征的综合视图,这可能影响跨类群的共翻译折叠过程。
总体而言,编码结构化(α螺旋和β折叠)区域和无序(无规则卷曲)区域的mRNA序列中的翻译速率模式在原核生物和真核生物中广泛观察到,这可能反映了进化的保守性和面临相同的进化压力。这种模式表明,mRNA序列中编码无序区域的翻译速率变慢可能确保结构化区域或结构域中的正确共翻译折叠有足够的时间。这一发现与先前的报告一致,即位于蛋白质结构域边界的核糖体停滞位点有助于和有利于共翻译折叠,从而避免潜在的错误折叠。
此外,rASA与翻译速率之间的关联模式在原核生物和真核生物中也是保守的,表明在所有测试的生物体中,编码暴露区域的RNA序列往往比编码埋藏区域的序列更慢地进行翻译,除了大鼠。此外,这种关联模式还观察到了翻译速率和局部绝对接触顺序之间。
在许多情况下,大鼠的关联模式与其他哺乳动物不同;例如,与其他哺乳动物相比,它在卷曲区域显示出较低的标准化印迹,而在α-螺旋和β-折叠区域则相反(图2J–L)。为了探索这些异常结果是否依赖于数据集,我们使用另一个数据集进行了额外的分析,分别来自大鼠(脑)、小鼠(肾)、大肠杆菌和人类(Hela细胞)(支持信息S2:表S2)。值得注意的是,为每个生物体选择的两个数据集来自不同的研究组。有趣的是,对于大多数蛋白质特征,包括蛋白质二级结构、脯氨酸残基、正电荷、IDR得分和局部绝对接触顺序,每个生物体的两个数据集之间的关联模式都是相似的(支持信息S1:图S6–S10)。这些结果表明,大鼠中的异常模式可能不是由于数据集特定的人为因素引起的,而可能是由于未知的固有因素引起的。一个可能的解释是高等真核生物基因表达更具组织特异性的性质。我们的Ribo-seq和蛋白质特征的综合分析需要具有高读取覆盖率(>60%)的基因,因此在高等真核生物中分析的基因数量相对较少(支持信息S2:表S4),这可能会影响每个数据集的结果。
在酵母中,带电的氨基酸可以强烈抑制翻译延伸速率。我们的分析在酵母和拟南芥中与先前的研究一致,显示出围绕带电氨基酸的标准版印迹峰,这表明带电氨基酸抑制酵母和拟南芥中的翻译。相反,我们的结果表明,在原核生物如大肠杆菌和铜绿假单胞菌中,正电荷可能会加速翻译(支持信息S1:图S4A,B)。可能的解释是原核生物和真核生物在出口通道几何形状上存在进化分歧,这是由于决定静电势的狭窄区域(出口通道的低谷)引起的。这种静电势的差异可能导致不同生物体中带电氨基酸残基与翻译速率之间的关联模式不同。
脯氨酸在原核生物和真核生物中都倾向于引起核糖体停滞,因为脯氨酸的氨基侧链已知在P位的肽键形成过程中既是差的A位受体又是差的P位供体。这可能是由于其有限的构象灵活性,阻碍了翻译延伸,正如在酵母、拟南芥和斑马鱼中报道的那样。此外,脯氨酸对核糖体停滞的影响可能是叠加的;在二脯氨酸基序列(XPPX)和多脯氨酸段(XPPPX)中,翻译停滞变得强烈。然而,在细菌中,脯氨酸基序列的核糖体停滞可以通过细菌中的翻译延伸因子EF‐P或真核生物中的起始因子IF5A得到救援,并且EF‐P的功能丧失将导致大肠杆菌中脯氨酸富集的基序中发生强烈的核糖体停滞。EF‐P和eIF5A都能增强核糖体的肽键形成酶活性,并促进脯氨酸等差的底物的反应性。在我们的结果中(支持信息S1:图S3),原核生物和真核生物中脯氨酸残基的效应是不同的,可能是由于解决脯氨酸引起的核糖体停滞的蛋白功能多样性(细菌中的EF‐P和真核生物中的IF5A)。
在这项研究中,我们仅探讨了翻译速率与蛋白质结构特征之间的关系。尽管如此,其他相关研究已经表明,蛋白质折叠速率与蛋白质结构(如蛋白质大小、形状、包装和横截面半径)也是相关的。此外,值得注意的是,表现出简单折叠动力学的蛋白质在细菌系统中相对于真核系统更容易折叠,这提供了关于蛋白质结构的进化分歧的线索。
结 论
总的来说,我们基于公开可用的Ribo‐seq数据进行了翻译速率与各种蛋白质特征的关联分析。我们揭示了翻译速率与蛋白质结构特征(二级结构元素、rASA和局部绝对接触顺序)之间的保守关系,表明不同生物体对蛋白质结构域施加不同的翻译速率以确保正确的蛋白质折叠。我们的研究还可以为增强重组蛋白表达提供新的思路。先前的研究仅考虑了mRNA序列特征,如密码子使用和mRNA二级结构来设计mRNA。我们的研究显示蛋白质结构也与翻译速率相关,表明蛋白质结构特征也可以纳入mRNA设计中,以精准调控共翻译折叠。我们的发现可能有助于生物制造领域的重组蛋白表达。
数据可用性声明
该流程现已在GitHub (https://github.com/ytksailab-org/VeloPro)上发布,文中所用的所有数据集也都上传到了线上的仓库中。补充材料(方法、图表、表格、脚本、图形摘要、幻灯片、视频、中文翻译版本和更新的材料)可以在在线DOI或iMeta Science(http://www.imeta.science/)中找到。
引文格式:
Bian, Bian, Toshitaka Kumagai, and Yutaka Saito. 2023. “VeloPro: A Pipeline Integrating Ribo‐seq and AlphaFold Deciphers Association Patterns Between Translation Velocity and Protein Structure Features.” iMeta 2, e148. https://doi.org/10.1002/imt2.148
作者简介

边遍(第一作者)
● 日本产业技术综合研究所人工智能研究中心助研,日本东京大学博士。
● 目前研究方向为深度学习,大语言模型,RNA的设计及优化,大规模多组学分析。

齐藤裕(通讯作者)
● 日本产业技术综合研究所人工智能研究中心主任研究员,日本东京大学客座副教授,北里大学客座教授,博士生导师。
● 研究方向为机器学习,生物大分子药物设计及优化,实验机器人开发及应用,生物信息学与大数据科学,已在Nucleic Acids Research、 Briefings in Bioinformatics、 ACS Catalysis、Bioinformatics、iMeta等期刊发表学术论文30余篇。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]