点击蓝字 关注我们
ViWrap:用于从宏基因组中识别、归类和预测病毒的宿主关系的模块化工具

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.118
● 2022年6月7日, 威斯康星大学周之超团队在 iMeta 在线发表了题为 “ViWrap: A modular pipeline to identify, bin, classify, and predict viral-host relationships for viruses from metagenomes ” 的文章。
● ViWrap的设计旨在便于广泛使用,以研究人类和环境系统中的病毒。ViWrap可通过GitHub(https://github.com/AnantharamanLab/ViWrap)公开获取。有关软件的详细描述、使用方法和结果解释可以在该网站上找到。
● 第一作者:周之超
● 通讯作者:Karthik Anantharaman ([email protected])
● 合作作者:Cody Martin, James C. Kosmopoulos

● 主要单位:美国威斯康星大学麦迪逊分校细菌学系
亮 点
● ViWrap整合了最先进的工具和数据库,用于对来自宏基因组和基因组的病毒进行全面的表征和研究。
● ViWrap提供了一个高度灵活、模块化、可定制和易于使用的流程,适用于各种应用和场景。
● ViWrap实现了标准化和可重复的病毒宏基因组学、基因组学、生态学和进化学的流程。
摘 要
病毒越来越被认识为人类和环境微生物组的重要组成部分。然而,微生物组中的病毒由于培养困难和缺乏足够的模型系统而难以研究。因此,从宏基因组中识别和分析未培养病毒基因组的计算方法引起了广泛关注。这样的生物信息学方法有助于从不同环境中产生的大规模测序数据集中筛选病毒。虽然已经开发了许多用于推动从宏基因组中研究病毒的工具和数据库,但缺乏集成工具,能够包括病毒研究的各个不同环节的全面工作流程和分析平台。在这里,我们开发了ViWrap,这是一个用Python编写的模块化流程。ViWrap将多种工具的功能整合到一个平台中,以实现病毒分析的各个步骤,包括识别、注释、基因组分组、物种和属水平聚类、分类学分配、宿主预测、基因组质量的表征、综合总结和直观的结果可视化。总体而言,ViWrap为来自宏基因组、病毒组和微生物基因组的病毒的广泛和严格的表征提供了一个标准化和可重复的流程。我们的方法具有使用多种不同选项进行各种应用和场景的灵活性,并且其模块化结构可以根据需要轻松添加其他功能。ViWrap的设计旨在便于广泛使用,以研究人类和环境系统中的病毒。ViWrap可通过GitHub(https://github.com/AnantharamanLab/ViWrap)公开获取。有关软件的详细描述、使用方法和结果解释可以在该网站上找到。
视频解读
Bilibili:https://www.bilibili.com/video/BV16k4y1p7T3/
Youtube:https://youtu.be/yZU21Yzynrw
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
不断增长的来自各种生态系统(包括自然环境、工业人工环境和人体微生物组相关环境)的宏基因组/病毒组装物库,为挖掘病毒多样性提供了宝贵的资源。自2016年以来,科学家们通过使用从宏基因组中获取的未培养病毒基因组丰富了公共数据库中的病毒收藏,并推动了我们对自然界中病毒的理解。这导致了重要的发现,即病毒在重塑微生物宿主代谢和推动全球生物地球化学循环方面起着重要作用。病毒编码辅助代谢基因(AMG),以增强宿主功能,通常是为了病毒自身的利益。这些AMG可以维持、推动或绕过重要的代谢步骤,并为病毒提供适应优势。鉴于许多未培养病毒基因组及其AMG的发现,科学家们揭示了它们在包括光合作用、甲烷氧化、硫氧化、氨氧化、氨化、碳水化合物降解等重要生态功能中的参与。尽管取得了这些进展,我们对病毒的理解仍然滞后于细菌和古菌,主要是由于缺乏用于研究和推进病毒生态学的可用工具。这需要更加关注开发计算技术,以便从微生物组中分析病毒,以及将研究重点放在宏基因组和宏转录组数据上。
通常从未培养的病毒基因组序列中恢复病毒的方法有两种,即从大量宏基因组或病毒组中恢复。大量宏基因组包括微生物群落的所有遗传物质,而病毒分数只占大量宏基因组的一小部分。另一方面,病毒组代表富集和浓缩的病毒分数,并排除了微生物群落的其他成员。已经开发了许多用于从大量宏基因组和病毒组中识别病毒的工具。例如,Microseek使用蛋白相似性来检测病毒。它通过对转录reads和contigs的最近共同祖先(LCA)与参考病毒蛋白数据库进行评分来实现这一目标。VIP将核苷酸和氨基酸序列与RefSeq参考数据库进行比对,以获得病毒reads,然后组装病毒contigs;此外,它还提供了分类鉴定、覆盖率和系统发育分析等下游分析。在所有当前可用的工具中,VIBRANT、VirSorter2和DeepVirFinder是用于识别大量宏基因组和病毒组中病毒的三个流行软件。VIBRANT使用混合机器学习和蛋白相似性方法来自动恢复和注释病毒。VirSorter2使用一组定制的自动分类器实现高病毒恢复性能。DeepVirFinder训练基于kmer的病毒机器学习分类器来识别病毒。
在病毒鉴定之后,已经开发了软件和方法来进行病毒基因组分箱、病毒分类学鉴定、基因组完成度估计和病毒宿主预测。vRhyme使用覆盖效应大小和病毒scaffolds的核苷酸特征来进行病毒基因组分箱。vConTACT2使用整个基因组基因共享网络进行基于距离的分层聚类和病毒分类学预测。dRep通过基于序列相似性来去重基因组,从而实现病毒聚类。CheckV用于检查病毒基因组的质量和完整性,iPHoP集成了目前所有可用的病毒-宿主关系预测方法,并构建了一个机器学习框架,以获取病毒的全面宿主预测。除了这些工具之外,多个先前建立的病毒数据库包含可用于指导病毒分类的蛋白序列。例如,NCBI RefSeq存储参考病毒基因组,VOGDB提供VOG HMM的预聚类病毒标记物(http://vogdb.org),IMG/VR v4数据库(目前最大的病毒特定基因组数据库)具有经过严格方法预分配的高质量病毒操作分类单元(vOTUs)。然而,虽然这些工具和数据库正被越来越多地使用,它们仅仅是在病毒多样性和生态学的全面分析的庞大链条中作为单独的链接。鉴于病毒学领域相对年轻,对于具有有限病毒和生物信息学技能的用户来说,往往很难知道使用哪些工具、如何整合方法和解释结果。一个集成的流程管线,涵盖病毒分析的整个工作流程,并提供易于阅读/解析的结果,将显著推动病毒学领域的发展,并使从宏基因组和微生物组中研究病毒的工作更加简易可复制。此外,这个集成的流程管线可以无缝链接到先进的下游病毒进化分析管线,例如MetaPop,从而增强对病毒基因组的进化动力学研究能力。
为了解决这个问题,我们开发了ViWrap,一个集成且用户友好的模块化流程管线,用于研究病毒多样性和生态学。ViWrap可以从宏基因组中识别、分组、分类和预测病毒的宿主关系。它集成了以下先进方法:1)在保持严格规则的同时对病毒进行全面筛选;2)一个标准化和可重复的流程管线,集成了先进的工具/数据库,并易于在将来添加额外功能;3)灵活的选项,用于识别方法,使用宏基因组reads(带有或不带有reads;短读长或长读长reads),以及针对不同应用场景的定制微生物基因组;4)一个一站式工作流程,用于生成易于阅读/解析的结果,并可视化和统计总结样本中的病毒。ViWrap将显著简化目前从宏基因组中研究病毒的计算程序,加快在新生成的或先前存储的宏基因组/病毒组中筛选更多的病毒多样性的研究,并促进对环境和人体微生物组中病毒群落结构和功能的理解。
方 法
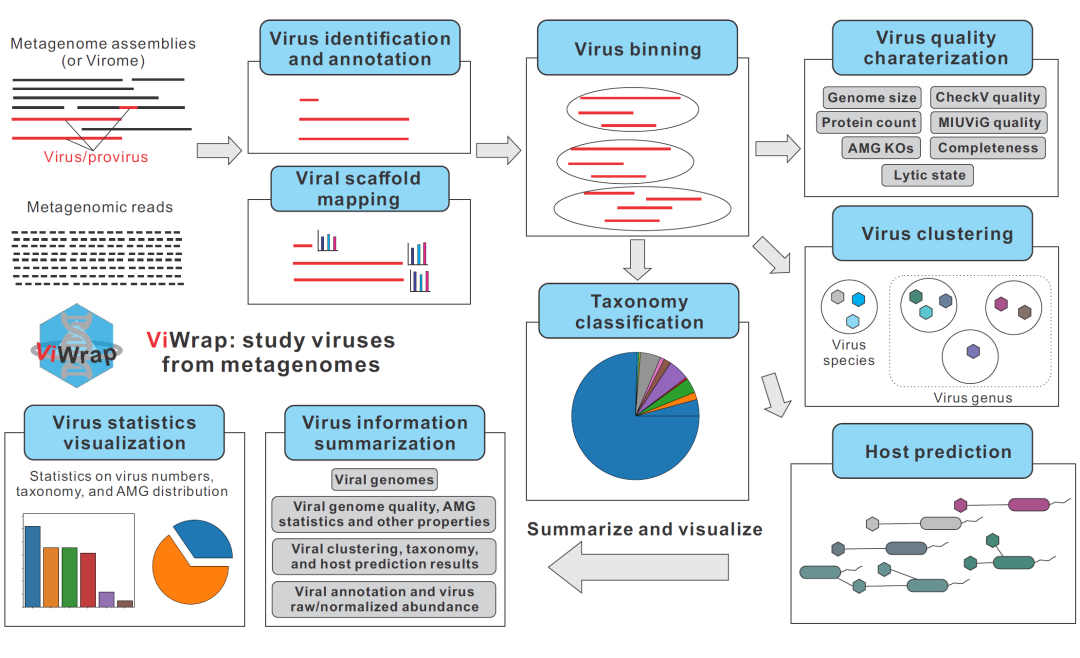
ViWrap是一个流程/封装程序,用于整合几个流行的病毒分析软件/工具,从宏基因组中识别、分组、分类和预测病毒与宿主的关系。它利用各种软件/工具将它们集成到一个模块化的流程管线中,以用户友好的方式获得有关病毒基因组学、生态学和多样性的综合信息。ViWrap具有八种不同的病毒分析功能,包括“病毒鉴定和注释”(通过VIBRANT、VirSorter2和DeepVirFinder)、“病毒分组”(通过vRhyme)、“病毒聚类”(通过vConTACT2到属级和dRep到种级)、“病毒分类学分类”(通过NCBI RefSeq病毒蛋白数据库、VOG HMM数据库和IMG/VR v4数据库)、“病毒信息总结”、“结果可视化”、“病毒质量表征”和“病毒宿主预测”(通过iPHoP)。预期的输入是宏基因组scaffolds或病毒组,以及宏基因组reads。
在这里,我们将宏基因组组装序列定义为从包含原核生物、真核生物和病毒混合群落的大量宏基因组中重建的scaffolds,将病毒组定义为从过滤/浓缩的病毒颗粒DNA中得到的组装序列,其中病毒占主导地位。宏基因组和病毒组的reads在本文其余部分统称为宏基因组reads。输出结果为用户友好的表格和图表,包括病毒基因组和相关统计数据、聚类、分类和宿主预测结果、注释和丰度结果,以及相应的统计总结的可视化(详见图1)。
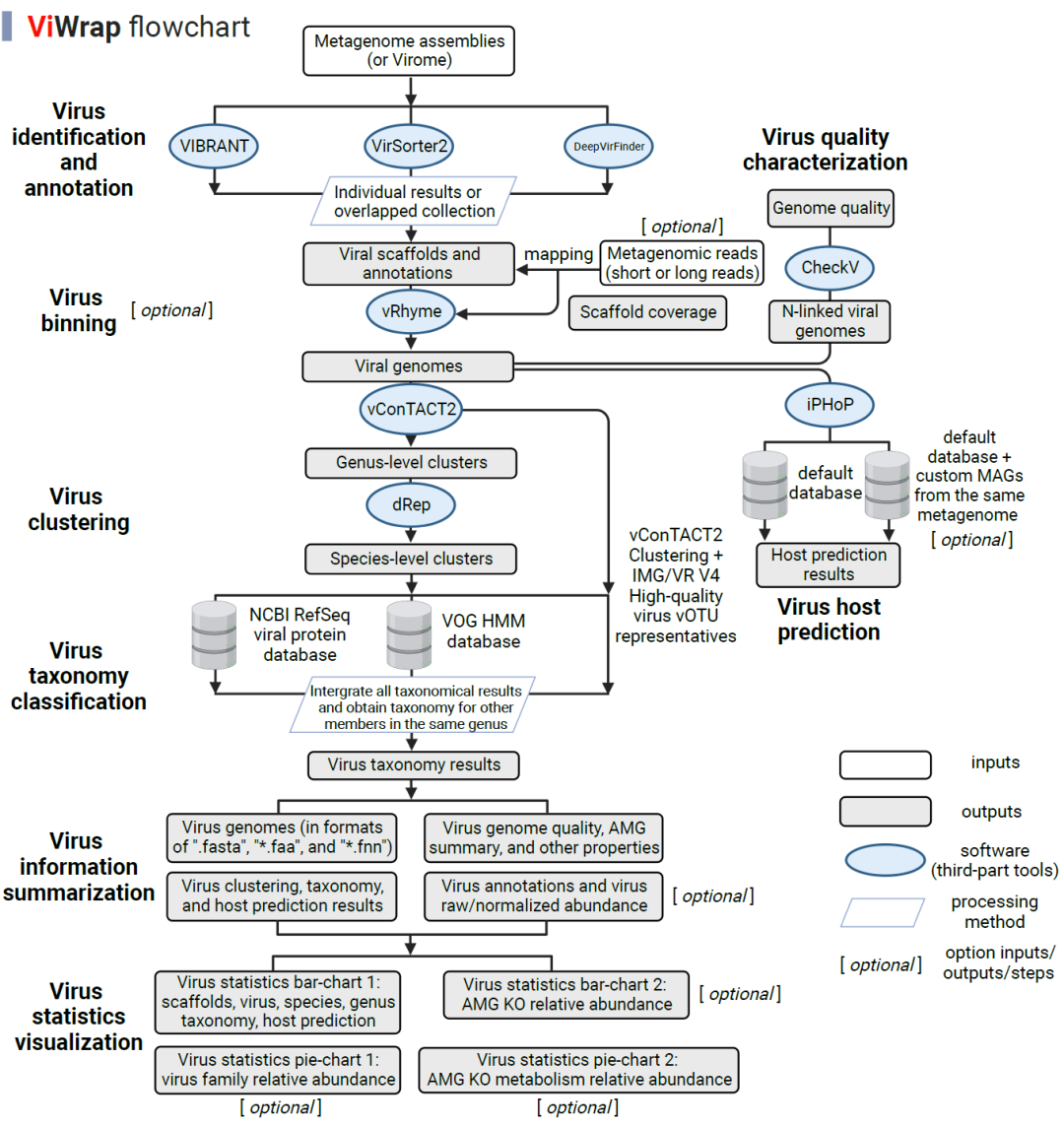
图1. 描述ViWrap中不同步骤和功能的流程图
空方块表示输入,填充的方块表示输出,椭圆表示软件,平行四边形表示用于获取下游结果的处理方法。
ViWrap可以与宏基因组reads一起使用或不使用,尽管使用reads可以带来优势并实现特定分析。具体来说,为了进一步方便使用宏基因组/病毒组/基因组进行病毒挖掘,但没有相应的宏基因组reads,我们引入了一个特定的“run_wo_reads” Python任务。ViWrap能够仅使用宏基因组/病毒组或基因组而不输入宏基因组reads。当应用此任务时,ViWrap将避免宏基因组mapping和病毒分组的步骤,因此仅报告单个scaffold分辨率下的病毒结果,而不包含基因组分组的结果。此外,我们实现了“set_up_env”和“download”任务,用于下载和设置conda环境和数据库的单一步骤。为了节省最终结果文件夹所需的存储空间,我们引入了一个“clean”任务,用于清除每个结果目录中的冗余信息。
ViWrap使用Python编写,需要conda环境才能实现适当的性能。该软件已存储在GitHub上(https://github.com/AnantharamanLab/ViWrap)。有关程序描述、安装、运行方法以及输入和输出解释的详细信息可以在GitHub页面上找到。我们使用了深海热液喷口环境(太平洋海域Guaymas盆地)中栖息的微生物群落的宏基因组组装序列和reads作为一个示例来运行ViWrap。为了便于用户使用此数据集进行测试,缩短运行时间,我们使用了一个子集的组装序列(18,000个scaffolds,占总数的约10%)和原始reads的两个子集,分别为总reads的10%和15%(随机选择)作为输入。此外,还使用了来自同一数据集的98个先前重建的宏基因组组装的基因组(MAGs),用于基于自定义宿主基因组的iPHoP的病毒宿主预测。
结 果
ViWrap工作流程
ViWrap的详细工作流程如图1所示。首先,ViWrap可以接受宏基因组或病毒组作为输入源来识别病毒。使用了三种方法来识别病毒scaffolds,分别是VIBRANT、VirSorter2和DeepVirFinder。可以根据用户选择的方法生成病毒识别的结果,可以是单个识别方法(即vb、vs或dvf)的单个结果,也可以是不同识别方法(即vb-vs或vb-vs-dvf)结果的交集。这三种方法在识别病毒方面具有不同的准确性和性能。根据使用这三种工具和其他七种工具对宏基因组中的病毒进行识别的研究的基准测试结果,这三种工具在处理人工RefSeq scaffolds时都表现出优秀的性能;精确度、召回率和F1得分也都超过0.85。使用基于基因的方法,VIBRANT和VirSorter2在广泛的病毒范围内具有高准确性,极大地促进了检测当前参考数据库中最常见病毒组之外的新病毒。VIBRANT使用基于机器学习的神经网络算法和定量的v-score度量来最大化高多样性病毒的识别。VirSorter2使用一组自定义的自动分类器来改善病毒的检测范围和准确性;它在最小化由真核基因组和质粒引入的分类错误方面具有高特异性。这两种工具都是病毒识别的最新工具。相比之下,DeepVirFinder目前是使用基于k-mer的方法预测病毒序列的最佳工具之一。然而,所有基于k-mer的方法都依赖于病毒参考数据库,这些数据库受限于对RefSeq/其他培养病毒的偏好,并且不考虑可以从宏基因组研究中恢复的不同病毒。因此,所有基于k-mer的方法都面临参考偏差的共同问题,这对于从宏基因组中识别新的和多样性病毒的算法有负面影响。因此,我们将“vb-vs”方法作为默认方法,生成一个全面而严格的病毒scaffolds集合,满足两种常用病毒识别方法的要求(图2)。这种“vb-vs”组合方法是推荐的设置。单独的病毒scaffolds识别方法的基准测试结果可以在先前的出版物中找到,但是在这里,我们不提供任何单个或组合的病毒scaffolds识别方法的基准测试结果。AMG(辅助代谢基因)的识别和病毒蛋白质注释是基于VIBRANT的结果进行解析的。具体来说,这里呈现的AMG识别是基于VIBRANT提供的AMG KO集合进行的,应该被视为初步结果。为了获得更可靠和准确的结果,需要进行手动验证。
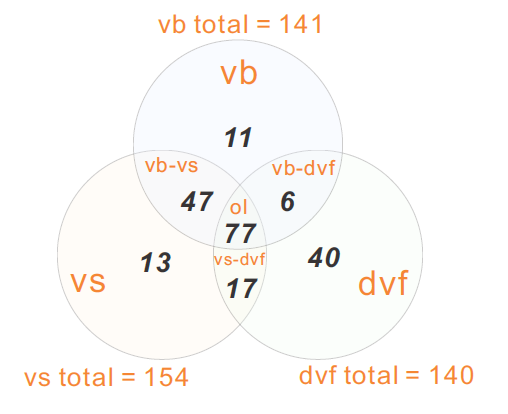
图2. Venn图表示三种方法鉴定出的重叠病毒scaffolds(交集)
缩写:“vb” - VIBRANT, “vs” - VirSorter2, “dvf” - DeepVirFinder, “vb-vs” - VIBRANT和VirSorter2, “vs-dvf” - VirSorter2和DeepVirFinder, “vb-dvf” - VIBRANT和DeepVirFinder, “ol” - “vb”,“vs”,和“dvf”之间的重叠病毒scaffolds。每个方法的结果来自于Guaymas Basin热液喷口样本的示例宏基因组数据集的演示。
在第二步中,使用宏基因组reads mapping到给定的宏基因组或病毒组以获取scaffolds覆盖度。可以使用短读长reads和长读长reads作为输入来生成scaffolds覆盖度结果。Scaffolds覆盖度文件用于通过vRhyme对病毒基因组进行分箱。为了将病毒scaffolds严格地分配到给定的病毒基因组中,我们采用了以下要求:1)在vRhyme设置中,病毒基因组的最大蛋白冗余设置为5;2)通过CheckV发现的“完整”病毒scaffolds不会被分配到病毒基因组中;3)具有一个或多个裂解成员和一个整合的原病毒的基因组分箱将不被考虑,并将被分割;4)具有两个或多个溶原成员(包括溶原scaffolds和整合的原病毒)的基因组分箱将不被考虑,并将被分割。最后,使用CheckV评估所有已识别的病毒的基因组质量。由于CheckV需要单个scaffold病毒作为输入,因此多个scaffold病毒基因组通过多个N进行连接以满足要求。然而,由于连接的顺序会影响ORF(开放阅读框)的预测,而且由于Prodigal在接近N连接处时对ORF预测的严格性,一些ORF可能无法被调用,因此这些通过N连接的多个fasta文件仅用于通过CheckV评估基因组质量。另外,CheckV无法检测来自不同病毒的污染,因此对于多个scaffold病毒分箱的完整性结果不能排除来自其他病毒物种的潜在污染。
在第三步中,通过vConTACT2对属水平聚类(病毒属)进行分类,同一“VC亚聚类”中的基因组被视为来自同一属。通过dRep对种水平聚类(病毒种)进行分类,ANI(平均核酸同源性)> 0.95且对齐分数 > 0.85的基因组被视为来自同一物种;请注意,此处描述的病毒物种可能不符合MIUiVG提出的“新参考物种”的定义。目前,vConTACT2尚未针对真核病毒进行测试或验证;建议仅在使用ViWrap时将其应用于原核宏基因组。
在第四步中,使用三种方法为每个病毒分配分类信息。其中两种方法包括使用NCBI RefSeq病毒蛋白质数据库进行蛋白质搜索,以及基于VOG数据库中的HMM标记蛋白质进行搜索,具体操作说明可以参考先前的描述。对于第三种方法,我们使用IMG/VR v4高质量vOTUs中的vOTU代表作为先前步骤中由vConTACT2分配的属水平聚类中的锚点来分配分类信息。最后,将这三种分类结果整合在一起。当一个病毒在这三种方法中有多个分类结果时,最终结果的优先顺序遵循NCBI RefSeq病毒蛋白质搜索方法、VOG HMM标记搜索方法和vConTACT2聚类方法。为了获得这三种方法都无法分配分类的病毒的分类信息,首先进入每个属中,确定是否已经使用NCBI RefSeq病毒蛋白质搜索方法对任何病毒基因组进行了分类(仅考虑此分类方法的命中结果),然后将分类信息扩展到属中的所有成员。
在第五和最后一步中,使用iPHoP预测病毒的宿主。可以使用默认的iPHoP数据库和来自同一宏基因组的自定义MAG来进行宿主预测。使用来自同一群落的自定义MAG可以有助于建立病毒和MAG之间的直接联系。
最后,呈现病毒信息,包括全面的病毒和AMG总结,并相应地可视化统计数据。
结果的布局
结果的文件夹和文件按照以下顺序排列在最终的输出目录中:
00_VIBRANT_VirSorter_input_metageome_stem_name:病毒识别步骤的结果。该文件夹包含VIBRANT和VirSorter2运行的结果文件夹;此外,还提供一个包含两次运行结果合并的文件夹。提供了病毒在合并结果中的注释文件,“fasta”(核苷酸序列)文件,“ffn”(基因序列)文件和“faa”(蛋白质序列)文件。
01_Mapping_result_outdir:reads mapping步骤的结果。文件夹中提供了CoverM(https://github.com/wwood/CoverM)生成的原始scaffolds覆盖度结果和作为vRhyme输入的转换后的覆盖度结果。
02_vRhyme_outdir:使用vRhyme进行基因组分箱的结果。该目录包含文件夹“vRhyme_best_bins_fasta”、“vRhyme_best_bins_fasta_modified”(通过上述严格标准进行修改的最佳分箱)和“vRhyme_unbinned_viral_gn_fasta”(作为单scaffold病毒处理的未分箱病毒scaffolds)。此外,还包含两个表格,表示“vRhyme_best_bins_fasta”文件夹中的病毒的裂解/溶原状态和基因组完整性信息。
03_vConTACT2_outdir:使用vConTACT2进行分类的结果。该目录包含病毒识别步骤中识别的病毒和IMG/VR v4高质量vOTUs的蛋白质和病毒聚类结果的组合。
04_Nlinked_viral_gn_dir:作为CheckV输入的N连接的病毒基因组。该目录包含具有多个N连接的所有scaffolds的病毒基因组。在这里,仅使用符合CheckV输入文件格式要求的单scaffold病毒(N连接或原始单scaffolds)。
05_CheckV_outdir:CheckV分析的结果。该目录包含每个病毒的单独CheckV结果文件夹和以每个病毒作为单个输入的汇总病毒基因组质量结果。
06_dRep_outdir:dRep聚类的结果。该目录包含分配给同一属的病毒物种聚类结果。
07_iPHoP_outdir:使用iPHoP进行宿主预测的结果。该目录包含使用默认iPHoP数据库和来自相同宏基因组的自定义MAGs的iPHoP结果文件夹(如果提供了自定义MAGs)。
08_ViWrap_summary_outdir:病毒的总结结果,包括“Genus_cluster_info.txt”(病毒属聚类),“Species_cluster_info.txt”(病毒种聚类),“Host_prediction_to_genome_m90.csv”(基因组水平的宿主预测结果;默认的置信度得分阈值为90),“Host_prediction_to_genus_m90.csv”(属水平的宿主预测结果;默认的置信度得分阈值为90),“Sample2read_info.txt”(reads计数和碱基数),“Tax_classification_result.txt”(病毒分类结果),“Virus_annotation_results.txt”(病毒注释结果),“Virus_genomes_files”(包含所有病毒基因组的“fasta”、“ffn”和“faa”文件),“AMG_results”(包含来自所有病毒基因组的AMG统计和蛋白质序列),“Virus_raw_abundance.txt”(原始病毒基因组丰度),“Virus_normalized_abundance.txt”(标准化病毒基因组丰度;以每个样本的100M reads为标准化单位)和“Virus_summary_info.txt”(所有病毒基因组的总结属性,包括基因组大小、scaffolds数目、蛋白质数量、AMG KOs、裂解/溶原状态、CheckV质量、MIUViG质量、完整性和完整性方法)。
09_Virus_statistics_visualization:病毒统计结果的可视化结果。该目录包含两个条形图和两个饼图。第一个条形图表示识别到的病毒scaffolds数目、病毒数目、病毒物种数目、病毒属数目、进行分类的病毒数目和预测到宿主的病毒数目。第二个条形图表示AMG KOs的相对丰度。第一个饼图表示病毒科的相对丰度。第二个饼图表示AMG KO metabolism的相对丰度。还提供了可视化的原始输入。
ViWrap_run.log:日志文件。该文件记录了发出的命令以及各个步骤和整个过程的时间记录。
通过对代表Guaymas盆地深海热液喷口宏基因组的测试数据集进行运行,我们从原始的18,000个宏基因组scaffolds中获得了124个病毒scaffolds,这些scaffolds被分成91个病毒。总运行时间约为14小时,使用Ubuntu 18.04.6 LTS(x86_64)服务器上的20个线程。在耗时最长的部分中,使用VIBRANT和VirSorter2从宏基因组中获得病毒scaffolds大约需要2小时,运行vConTACT2对病毒基因组进行聚类大约需要45分钟,使用默认数据库进行iPHoP宿主预测大约需要30分钟,而使用自定义MAGs作为数据库则需要约10小时(创建新数据库的时间较长,因为此过程受限于iPHoP中实现的系统发育树构建方法)。
基于病毒统计的可视化结果通常表示病毒数目、分类和AMG分布的发现(图3)。从124个病毒scaffolds中,我们重构了91个病毒基因组(包括分箱和未分箱的病毒)(图3A)。每个病毒基因组属于一个不同的物种,并进一步分类为81个病毒属(图3A)。在91个病毒基因组中,有27个具有分类,8个具有宿主预测(图3A)。在分类方面,分配了三个科和一个纲,其总病毒相对丰度约为29.7%(图3B)。在病毒群落中发现了23个AMG KOs,并分配了相应的相对丰度比例(图3C)。当将KOs分类为KEGG metabolisms时,发现了两种代谢,即碳水化合物代谢和辅助因子和维生素代谢,它们占据了整个比例(图3D)。可视化结果提供了病毒群落的常规定量特征的直观和有用的解释,包括病毒数目和统计数据、病毒科相对丰度、AMG KO相对丰度和AMG KO代谢相对丰度(图3)。
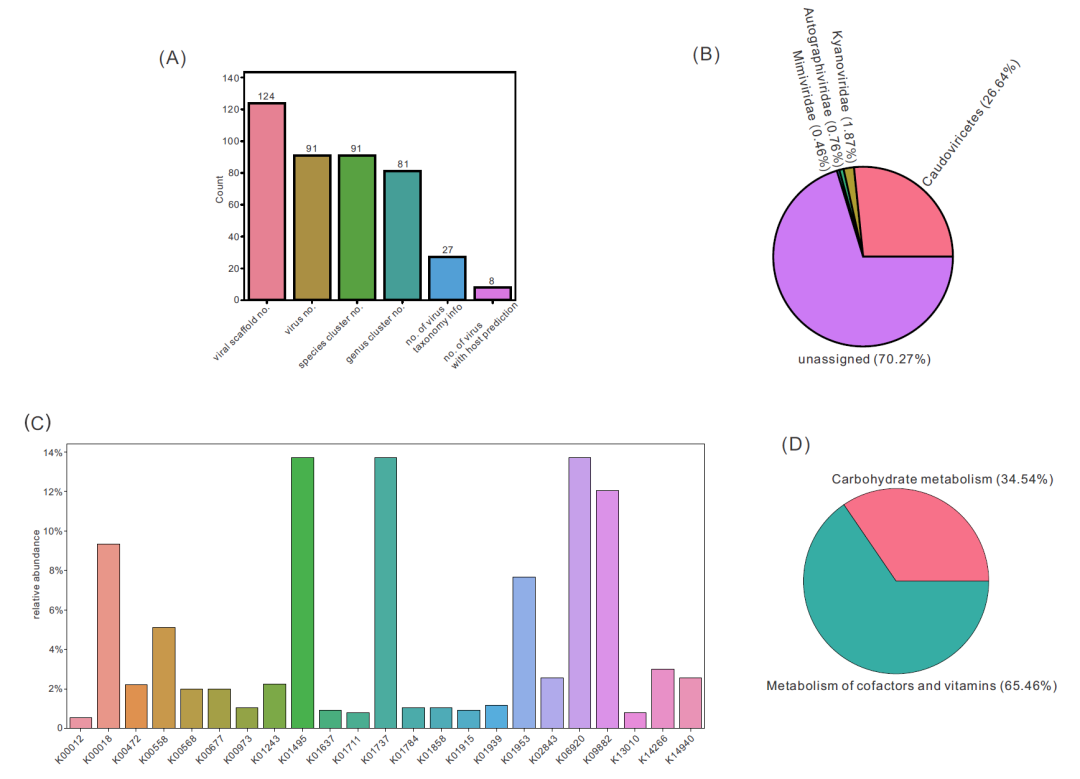
图3. 病毒统计数据的可视化结果
(A)柱状图表示鉴定出的病毒scaffolds数目、病毒数目、病毒物种数目、病毒属数目、具有分类学归属的病毒数目和具有宿主预测的病毒数目。(B)饼图表示病毒科的相对丰度。(C)柱状图表示AMG KO的相对丰度。(D)饼图表示AMG KO代谢的相对丰度。
讨 论
ViWrap是一个模块化且综合的流程,集成了完整的病毒分析软件/工具链。与之前只处理病毒分析的特定步骤(如病毒鉴定或病毒分组)的软件和工具不同,ViWrap对于解释病毒多样性和生态学的整体“链条”非常关键。与仅进行完整分析中的特定“环节”相比,一个集成的工具现在对于解释病毒多样性和生态学至关重要。显著的是,ViWrap减轻了用户在进行分析时选择合适的软件/工具的负担。随着对来自宏基因组的非培养病毒基因组研究的重要性日益增加,ViWrap的标准化方法将使用户能够以用户友好的方式从宏基因组中鉴定和分析病毒。
ViWrap集成了许多先进的主流和热门的病毒分析软件/工具。它利用这些组件工具实现了对宏基因组中病毒的全面筛选。ViWrap将病毒分析流程中每个步骤的输入和输出无缝连接,并预先配置了所有设置。这个特性节省了用户的时间,因为他们可能没有必要的知识来选择分析中每个工具的合适选项和参数。ViWrap还整合了上游的结果,并将其用于下游分析,无需重新进行相同的分析。例如,我们在病毒聚类和病毒分类两个步骤中都使用了vConTACT2聚类分析。通过将参考vOTU病毒(属代表性病毒)与查询病毒放在一起,并同时进行vConTACT2聚类,用户将通过整合两个步骤的分析节省大量时间。
该软件为用户提供了灵活的自定义选项,包括选择鉴定方法、利用宏基因组reads(包括长读长和短读长reads)以及将来自相同宏基因组的自定义MAG作为额外的宿主预测数据库。因此,它适用于各种应用场景,例如揭示微生物组或环境中的病毒多样性和生态学、在宏基因组中识别病毒和噬菌体、在基因组reads不可获取时从公共可用基因组中识别潜在病毒、发现与来自同一宏基因组的MAG重建的病毒之间的直接联系等。由于病毒序列通常较短,因此在病毒测序和分析中对长读测序技术的需求越来越大。ViWrap利用长读测序数据生成覆盖率,非常适合这些应用,因此有助于未来的病毒测序和分析。ViWrap还提供了全面的病毒分析结果和可视化统计数据,可以轻松用于进一步的下游分析和结果解释。ViWrap提供的统计总结全面展示了系统中的病毒群落和病毒生态功能。
总之,ViWrap是一个一站式模块化流程和封装程序,将宏基因组/病毒组和/或宏基因组reads作为输入,以用户友好、全面、标准化(对于各种应用场景灵活)的方式生成易于阅读/解析的病毒分析结果。尽管我们在自然环境(本研究中的热液喷口环境)中展示了ViWrap的应用,但在ViWrap中实施的工具和数据库使其能够广泛应用于各种环境,例如人造环境(工业环境、废水处理厂)和与人类微生物组相关的环境(人体、人体消化道、口腔)。随着微生物组中病毒和噬菌体领域的快速发展,越来越多的大型数据集和更先进的软件/工具正在被开发和引入。ViWrap的模块化性质将确保在将来轻松集成新的工具和数据库。我们认为ViWrap有潜力在学术界广泛应用,并标准化和推进微生物组中的病毒研究。
代码和数据可用性:
来自Guaymas Basin热液喷口环境的示例宏基因组数据集的重构基因组和宏基因组reads可在NCBI BioProject PRJNA522654和SRA SRR3577362上获得。ViWrap公开在GitHub上(https://github.com/AnantharamanLab/ViWrap)供所有研究人员使用,并提供详细的说明。补充资料(图表、脚本、图形摘要、幻灯片、视频、中文翻译版本和更新材料)可在在线DOI或iMeta Science(http://www.imeta.science/)上找到。
引文格式:
Zhichao Zhou, Cody Martin, James C. Kosmopoulos, Karthik Anantharaman. ViWrap: A modular pipeline to identify, bin, classify, and predict viral-host relationships for viruses from metagenomes. iMeta 2023. https://doi.org/10.1002/imt2.118
作者简介

周之超(第一作者)
● 美国威斯康星大学麦迪逊分校细菌学系助理科学家。
● 研究方向为深海热液微生物组及病毒组。本科就读于武汉大学生物学基地班(2006-2010),期间发表1篇一作SCI论文(导师:彭方教授),获得国家海洋局2012年度极地科学优秀论文三等奖;博士期间在香港大学顾继东教授实验室从事海洋微生物分子生态学研究,获得University Postgraduate Fellowship奖励(每年每学院仅一人)。2016年作为亚洲唯一一名学生受Lorus J. & Margery J. Milne Scholarship资助参加国际著名的伍兹霍尔海洋生物实验室的Microbial Diversity夏季课程(课程主任为加州理工学院著名环境微生物科学家Jared Leadbetter和美国科学院院士Dianne Newman)。博士毕业后在香港中文大学罗海伟教授实验室研究近海湿地微生物进化,强化了分子进化和生信技能。2019年至今在美国威斯康星大学细菌学系Karthik Anantharaman教授实验室继续从事深海热液微生物组及病毒组研究。2023年获得Diversity 2023 Young Investigator Award。近几年,在微生物组学分析方法、微生物碳和硫循环等取得了一系列成果,共发表40+篇SCI论文(一作/共一20+篇),包括发表在The ISME Journal (4), Nature Communications(1共一), Microbiome (1共一,1一作)等著名杂志,文章总引用数2301次,H-index为24(Google Scholar)。个人网站:https://zczhou2017.wixsite.com/mysite。

Karthik Anantharaman
(通讯作者)
● 美国威斯康星大学麦迪逊分校细菌学系副教授。
● 研究方向为微生物和病毒生态学。Anantharaman在印度孟买长大,并于2007年在印度国立技术学院卡纳塔克那里获得土木工程学士学位。随后,他于2014年在密歇根大学Gregory Dick教授的指导下获得地球与环境科学博士学位,研究热液喷口的微生物学。在加利福尼亚大学伯克利分校与Jillian Banfield教授的博士后培训期间,他利用高分辨率宏基因组学研究了陆地地下的微生物生物地球化学。Anantharaman荣获多项奖励,包括NSF职业生涯奖、NIH杰出研究员(早期研究者-MIRA)奖、美国微生物学会早期职业环境研究奖,卡夫里科学前沿研究学者奖,Vilas 早期研究者奖。Anantharaman的跨学科研究项目结合了计算、实验室和野外实验的方法,以理解海洋和淡水环境以及人类健康中的微生物和病毒过程对生物地球化学转化的影响。随着越来越多人认识到病毒和噬菌体是所有微生物组的组成部分,Anantharaman及其团队正在开发和应用最先进的计算方法和模型系统,以研究自然界中的病毒生态学和相互作用。实验室网站:https://anantharamanlab.wisc.edu/。
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]