点击蓝字 关注我们
DeepKla:基于注意力机制的蛋白质赖氨酸乳酸化位点预测工具

https://doi.org/10.1002/imt2.11

4.5
iMeta
SHORT COMMUNICATION
● 2022年3月15日,电子科技大学吕昊等在iMeta在线发表题为“DeepKla: An attention mechanism-based deep neural network for protein lysine lactylation site prediction”的方法类文章。
● 该研究提出第一个用于识别蛋白质赖氨酸乳酸化位点的计算工具DeepKla,实验结果证明 DeepKla 具有出色的预测能力和鲁棒性。并搭建了一个在线服务器,基于所提出的模型建立了一个名为DeepKla 的在线服务。
● 第一作者:吕昊

● 通讯作者:林昊(电子科技大学信息生物学中心)
● 合作作者:刀福英
摘 要
作为一种新型的蛋白质翻译后修饰,赖氨酸乳酸化(Kla)在各种细胞过程中起着举足轻重的作用。高通量质谱法是检测 Kla 位点的主要方法。然而,与计算方法相比,识别 Kla 位点的实验方法通常既费时又费力。因此,需要开发一种强大的工具来识别 Kla 位点。为此,我们通过结合有监督的嵌入层、卷积神经网络、双向门控循环单元和注意力机制层,提出了DeepKla 的计算框架,用于识别水稻中的 Kla 位点。综合实验结果证明 DeepKla 具有出色的预测能力和鲁棒性。基于所提出的模型,我们建立了一个名为 DeepKla 的在线服务,可在 http://lin-group.cn/server/DeepKla 上免费访问。DeepKla 算法的源码可在GitHub (https://github.com/linDing-group/DeepKla) 获取。
亮 点
● 提出第一个用于识别水稻中的 Kla 位点的计算工具,DeepKla
● 使用有监督的嵌入层、卷积神经网络、双向门控循环单元和注意力机制层构建模型
● 建立用户友好的在线服务,DeepKla (http://lin-group.cn/server/DeepKla)
视频解读
Bilibili:https://www.bilibili.com/video/BV1BL4y157U9/
Youtube:https://youtu.be/_tr6QefWcnc
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
赖氨酸乳酸化(Kla)是一种存在于哺乳动物、植物和真菌细胞中的新型翻译后修饰(PTM) [1-3]。生化上,Kla在赖氨酸残基的e胺基上引入了一个小的乳酸基团,质量为72.021道尔顿 [4]。越来越多的证据表明,乳酸化与炎症反应 [3, 5]、肺纤维化的进展 [6] 和细胞重编程相关 [7]。然而,Kla在影响细胞进程中的调节作用仍不清楚。
Kla位点的常规表征方法是一种基于质量偏移的高效液相色谱-串联质谱(MS/MS)技术 [4]。然而,湿实验的固有缺陷导致全蛋白组识别Kla较为困难。因此,需要计算方法来填补湿实验的空白。
据我们所知,目前尚未有水稻中Kla位点的计算预测模型发表。因此,在本研究中,我们提出了一种新颖的基于深度学习的模型,名为DeepKla,用于准确识别蛋白质乳酸化位点。作为一个集成的深度学习框架,DeepKla由四个紧密连接的子网络组成,包括词嵌入层、卷积神经网络(CNN)、双向门控循环单元(BiGRU)和注意力机制层。具体来说,嵌入层使用蛋白质序列作为唯一输入自动提取序列特征,从而避免人为设计导致的有偏差的特征。此外,BiGRU和注意力机制分别用于从蛋白质序列中捕获远程信息和关键位置信息。基准实验结果表明,嵌入层和CNN-BiGRU-注意力机制层生成的鲁棒表征在识别Kla位点上具有很强的预测性能。我们认为,该框架还能够适用于解决其他PTM位点识别问题。
方 法
基准数据集
在本研究中,水稻乳酸化数据从文献中获取 [2]。赖氨酸(K)上的有注释的乳酸化位点被用作正样本,而相同的氨基酸排除来自相同蛋白质的注释的乳酸化位点被用作负样本。经过预评估不同窗口大小的表现,当窗口大小为51时能够最大限度地提取Kla位点信息。此外,我们使用CD-HIT程序 [8] 并设置相似性阈值为30%去除冗余数据。由于正负样本极不平衡,因此我们采用将正样本过采样的方法保持正负样本的比例为1:1。另外,我们从文献 [1] 中收集了273个灰霉菌中的Kla数据作为测试数据,以客观地评估模型。数据的详细信息见表1。
表1 本研究中使用的训练集和独立集
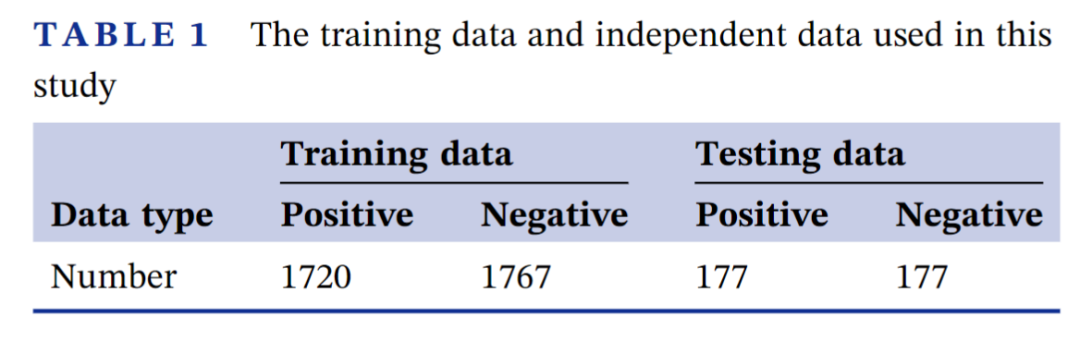
DeepKla的序列表征和框架
图1展示了我们用于Kla位点预测的深度学习框架。对特定的蛋白质序列,我们使用有监督的嵌入层对其进行编码,该嵌入层已成功应用于PTM位点预测问题 [9, 10]。在DeepKla的深度学习框架中,多层CNN将输入的蛋白质序列编码为固定的二维隐藏状态,随后,将二维隐藏状态输入至BiGRU。除此之外,注意力机制层被用于捕捉蛋白质序列的位置信息。序列表征和算法框架设计的详细描述见补充文件。

图1 DeepKla流程图
该模型基于Keras(2.0.6版)实现,并于1.4 GHz Intel Quad-Core i5 的 MacOS 上执行。我们在训练过程中为RMSProp优化器设置了默认的学习率,并使用64作为批次大小。模型结构和训练数据的超参数基于五倍交叉检验确定。为了避免过拟合,我们设置了早停机制
结 论
DeepKla的工作流如图1所示。在数据收集和预处理之后,DeepKla为每个蛋白质序列中的氨基酸分配整数。CNN-BiGRU-注意力机制层不断迭代以捕获蛋白质序列的特征。在输出层,使用全连接层和softmax生成预测结果。
模型性能评估
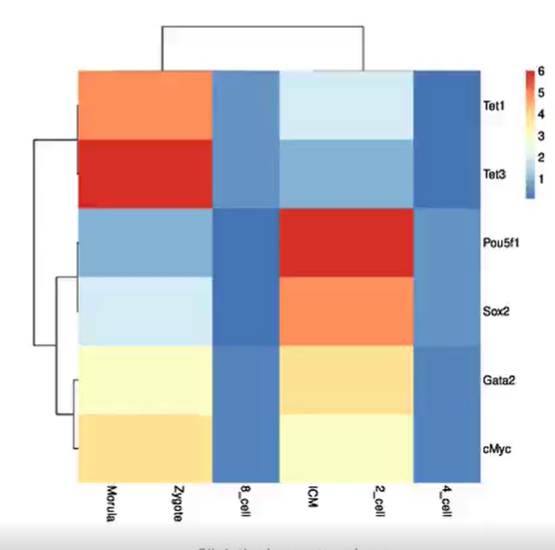
为了评估DeepKla的预测性能,我们进行了五折交叉检验。为此,我们将训练数据随机划分为五个不重叠的子集。在每个验证步骤中,五分之四的数据用于训练模型,其余五分之一的数据用于测试其性能。五种评价指标包括敏感性(Sn)、特异性(Sp)、精确度(Acc)、马修斯相关系数(MCC)和ROC曲线下面积(AUC)绘制在图2A、C中。结果表明DeepKla可以产生0.9901的AUC(图2C),证明了DeepKla在识别Kla位点问题中的稳定性。
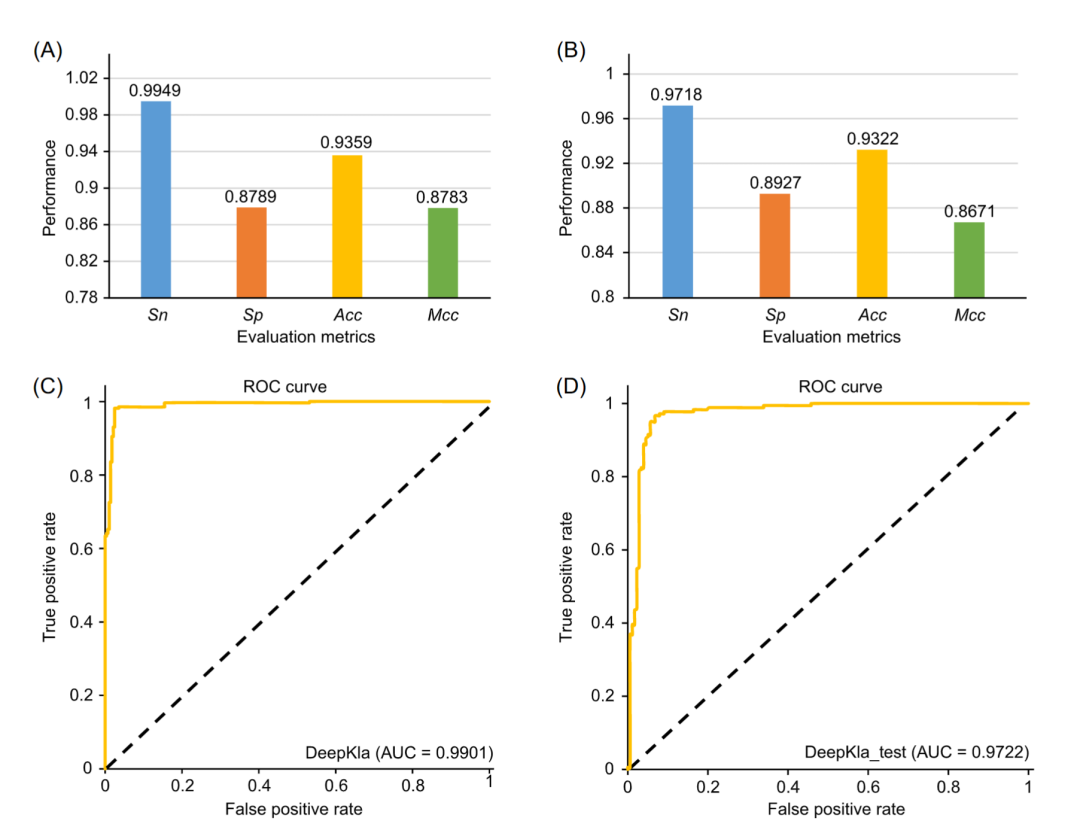
图2 基于训练集和独立集的预测指标
(A, B)分别为训练集(A)和独立集(B)的预测指标。(C, D)分别为训练集和独立集的ROC曲线。模型性能评价指标定义为Sn(敏感性)= TP/(TP+FN),Sp(特异性)= TN/(TN+FP),Acc(精确度)= (Sn+Sp)/2,MCC(马修斯相关系数)= (TP*TN-FP*FN)Ö(TP+FP)(TP+FN)(TN+FP)(TN+FN),其中TP=真阳性,FP=假阳性,TN=真阴性,FN=假阴性。
独立集已经被广泛用于评估分类器的稳定性。因此,我们从文献中 [1] 收集了灰霉菌中的273个Kla数据,以进一步评估DeepKla的性能。在经过与基准数据集相同标准的处理之后,获得了177个Kla序列和177个非Kla序列。如图2B、D所示,DeepKla始终能够产生令人满意的性能(Sn: 0.9718, Sp: 0.8927, Acc: 0.9322, MCC: 0.8671, AUC: 0.9722)。这一结果表明,DeepKla在识别Kla位点方面具有出色的预测能力和可迁移性。
我们评估了影响DeepKla性能的不同策略的贡献。通过在相同独立集上进行测试,我们比较了是否添加注意力机制层对DeepKla性能的影响。结果表明,包含注意力机制的框架获得了更好的性能(精确度=94.07% [333/354]),而没有注意力机制的框架的表现为92.09%(326/354)。这一结果突出了注意力机制的有效性,该机制能够捕获Kla预测问题中的关键信息。我们还分别比较了DeepKla在CNN-BiGRU-注意力机制层和CNN-BiLSTM-注意力机制层的性能。结果表明,包含BiLSTM的框架性能较弱(精确度=85.59% [303/354]),表明BiLSTM的改进版本,即BiGRU,在提高Kla位点的预测能力上更具优势。
DeepKla在线服务
为了方便使用,我们搭建了一个在线服务器。Web服务器只接受FASTA格式的蛋白质序列。服务器设置了两种输入方式,一种是直接将要预测的序列粘贴到空白框中,另一种可将问题序列以问价形式上传。需要注意的是,问题序列中不能包含“X”等特殊字符,否则模型将无法识别并返回错误报告。预测完成后,预测结果会以表格形式展示在结果界面。

http://lin-group.cn/server/DeepKla/
引文格式:Hao Lv, Fu-Ying Dao, Hao Lin. 2022. DeepKla: An attention mechanism-based deep neural network for protein lysine lactylation site prediction. iMeta 1: e11. https://doi.org/10.1002/imt2.11
参考文献
1. Gao M, Zhang N, Liang W. Systematic Analysis of Lysine Lactylation in the Plant Fungal Pathogen Botrytis cinerea, Front Microbiol 2020;11:594743.
2. Meng X, Baine JM, Yan T et al. Comprehensive Analysis of Lysine Lactylation in Rice (Oryza sativa) Grains, J Agric Food Chem 2021;69:8287-8297.
3. Zhang D, Tang Z, Huang H et al. Metabolic regulation of gene expression by histone lactylation, Nature 2019;574:575-580.
4. Yu H, Bu C, Liu Y et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair, Sci Adv 2020;6:eaay4697.
5. Irizarry-Caro RA, McDaniel MM, Overcast GR et al. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation, Proc Natl Acad Sci U S A 2020;117:30628-30638.
6. Cui H, Xie N, Banerjee S et al. Lung Myofibroblasts Promote Macrophage Profibrotic Activity through Lactate-induced Histone Lactylation, Am J Respir Cell Mol Biol 2021;64:115-125.
7. Li L, Chen K, Wang T et al. Author Correction: Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade, Nat Metab 2020;2:1179.
8. Fu L, Niu B, Zhu Z et al. CD-HIT: accelerated for clustering the next-generation sequencing data, Bioinformatics 2012;28:3150-3152.
9. Lv H, Dao FY, Zulfiqar H et al. DeepIPs: comprehensive assessment and computational identification of phosphorylation sites of SARS-CoV-2 infection using a deep learning-based approach, Brief Bioinform 2021;22.
10. Lv H, Dao FY, Guan ZX et al. Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method, Brief Bioinform 2021;22.
作者简介

吕昊(第一作者)
● 电子科技大学博士研究生,苏黎世大学联合培养博士研究生
● 研究方向主要有利用机器学习/深度学习算法识别表观遗传修饰位点,以及利用单细胞三维基因组数据对细胞类型注释等

刀福英(合作作者)
● 电子科技大学博士研究生,南洋理工大学联合培养博士研究生
● 研究方向主要有基于机器学习方法识别真核基因组复制起始位起始位点以及三维基因组染色质互作的识别与应用

林昊(通讯作者)
● 电子科技大学信息生物学中心教授,科睿唯安2018年全球高被引科学家,爱思唯尔2020年中国高被引科学家,斯坦福大学2021年全球前2%科学家
● 曾先后获得四川省优秀青年人才(2019),四川省(2012)、河北省(2015)科技进步奖等。主要研究方向为大分子数据的表示、临床数据分析和疾病风险预测等
更多推荐
(▼ 点击跳转)
iMeta | 南昌大学丁霞等-水产养殖系统对中华鳖微生物组和肠道代谢组的影响
iMeta | 华中科大宁康组综述用于蛋白质结构预测的宏基因组定量分析
iMeta | 中科院李小方等膳食甘草促进小鼠镉解毒并调节肠道菌群代谢
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]
微信公众号
iMeta
责任编辑
微微