点击蓝字 关注我们
免疫相关相互作用扰动网络揭示胶质母细胞瘤的生物学特性和临床意义

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.127
● 2023年7月16日,郑州大学韩新巍、张振宇和刘灶渠团队在 iMeta 在线联合发表了题为 “Immune-related interaction perturbation networks unravel biological peculiars and clinical significance of glioblastom” 的文章。
● 本研究从免疫相关相互作用扰动网络的角度进行分析,对推动胶质母细胞瘤的个体化治疗起到重要作用。
● 第一作者:刘灶渠、徐雨迪、王宇慧
● 通讯作者:韩新巍 ([email protected])、张振宇 ([email protected])、刘龙([email protected])
● 合作作者:翁思远、徐辉、任雨晴、郭春光
● 主要单位:郑州大学第一附属医院介入放射科、介入研究所;河南省介入诊疗与临床研究中心;郑州大学第一附属医院神经内科、第三附属医院临床检验科、第一附属医院呼吸与危重症医学科、第一附属医院血管外科、第一附属医院肝胆胰外科、第一附属医院神经外科
亮 点
● 该研究引入了免疫相关相互作用扰动网络框架,该框架既考虑了生物网络中重要的相互作用信息,又体现了免疫与癌症之间的关系;
● 基于免疫相关相互作用扰动网络,识别出了四种具有不同临床结果和生物学特征的胶质母细胞瘤亚型,并在外部队列和内部队列中得到了验证;
● 该分型为理解胶质母细胞瘤异质性和促进个体化治疗提供了前景。
摘 要
免疫系统是一个由大量分子组成的相互作用网络,利用这个网络可以更好地描述免疫与癌症之间的关系。本研究旨在研究免疫相关相互作用扰动网络在胶质母细胞瘤中的行为模式。利用TCGA/CGGA胶质母细胞瘤组织和GTEx正常脑组织的RNA-seq数据,引入免疫相关相互作用扰动框架来表征四种不同亚型。四个亚型的稳定性和鲁棒性在公共数据集和我们的内部队列中得到了验证。在四个亚型中,C1是炎性亚型,具有高免疫浸润、低肿瘤纯度和对免疫治疗的潜在反应;C2是侵袭性亚型,特征是预后不良、端粒酶逆转录酶启动子突变、具有中等水平的免疫和基质成分,对受体酪氨酸激酶信号抑制剂敏感;C3为增殖亚型,肿瘤纯度高,免疫浸润水平低,对磷脂酰肌醇3'-激酶信号转导抑制剂和DNA复制抑制剂敏感,对免疫治疗不应答;C4为突触发生亚型,预后最好,高表达突触发生相关基因,异柠檬酸脱氢酶突变水平高,对放化疗具有潜在的敏感性。总之,该研究从免疫相关相互作用扰动网络的角度进行分析,对推动胶质母细胞瘤的个体化治疗起到重要作用。
视频解读
Bilibili:https://www.bilibili.com/video/BV1ZV41157k4/
Youtube:https://youtu.be/VyUPFhRl6T8
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
根据2021年世界卫生组织(WHO)对中枢神经系统(CNS)肿瘤的分类,胶质母细胞瘤(GBM)被重新定义为异柠檬酸脱氢酶(IDH)野生型胶质母细胞瘤。GBM具有浸润性生长模式和难治性。对于GBM,手术切除辅以替莫唑胺(TMZ)和同步放疗是标准常规治疗方法,但仍然效果不佳。肿瘤的异质性可能是GBM治疗的有限效果和快速进展的一个原因。基于基因表达谱的亚型探索在过去十年中已经普遍存在。分子分类的最新进展为GBM的异质性提供了关键见解,并促进了个体化治疗。然而,常规进行的bulk-RNA测序是在特定的时间或条件下进行的,忽略了生物系统是动态变化的事实。相反,包含基因和相互作用信息的生物网络在时间和条件上相对稳定。先前有研究利用基因相互作用扰动网络确定了乳腺癌的四个稳健亚型。此外,近期研究表明,相互作用生物标志物可以作为区分疾病或表型的有效可靠工具。因此,从基因相互作用网络的角度进行亚型识别可以为GBM的生物学意义和临床管理提供新的见解。
由众多免疫分子和细胞相互作用形成的免疫网络在各种生物过程中起着关键作用,尤其是肿瘤。先前的研究表明,免疫网络与癌症的发生和进展密切相关,能够预测肿瘤患者的预后并指导治疗。侯等人已经证明,免疫调节相互作用网络与GBM患者不良预后密切相关。GBM中肿瘤细胞与免疫微环境之间的相互作用最终抑制了分子途径的有益模式,增强了其恶性程度并提高了对癌症治疗的抵抗力。因此,基于免疫网络相互作用扰动来理解肿瘤异质性至关重要。
从免疫相关相互作用扰动网络的角度来看,本研究发现并验证了四种具有不同生物学特性和临床预后的亚型。这种分类方法为解读胶质母细胞瘤异质性并促进个体化管理提供了前景。
结 果
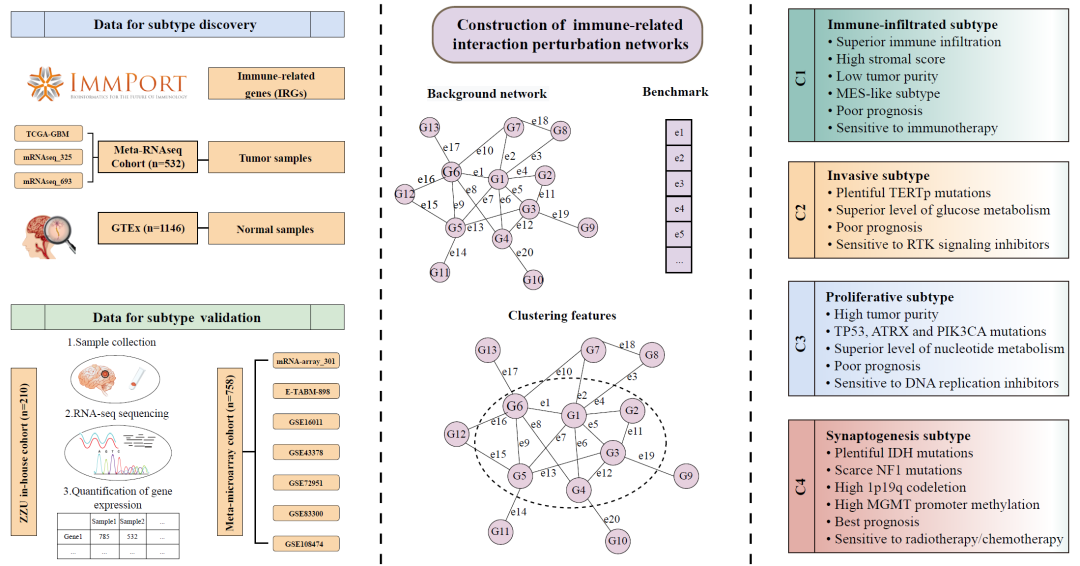
利用免疫相关边扰动矩阵构建四种亚型
为了生成免疫相关基因(IRGs)的相互作用扰动矩阵,本研究采用了先前报道的流程。通过STRING工具获得IRGs的蛋白质相互作用,将置信度大于0.7的基因对纳入背景网络中(图1和图S1)。该网络由1264个节点和15,347条边组成(表S1和表S2)。该网络是无标度网络,只有少数节点显示中心连接性(图S2A)。如先前报道的,从大样本量的队列中识别出小部分潜在亚型的可能性更大。此外,TCGA和CGGA数据库中的RNA-seq数据与GTEx队列(RNA-seq数据)兼容。因此,将Meta-RNAseq队列(通过ComBat算法去除了批次效应)作为GBM样本输入,将GTEx队列作为正常样本输入。正如在方法中提到的,首先通过对每个样本中每个基因的表达进行排序,得到表达秩矩阵;然后根据背景网络相互作用将其转换为差值秩矩阵(图1)。先前的研究表明,正常组织中的基因相互作用比肿瘤组织中更稳定和保守。因此,我们将所有正常样本的平均差值秩向量作为基准,生成相互作用扰动矩阵(图1)。该矩阵可以反映单个样本的相互作用扰动。正如预期的那样,肿瘤样本显示比正常样本更强的扰动(图S2B)。GBM样本中扰动的平均绝对值大小为81.4,是正常样本的两倍以上。随后,我们随机选择了3000个特征,并观察到GBM样本相对于正常样本显示出更大的变异和更高的扰动(图S2B)。以上结果表明,相互作用扰动可以代表样本的病变程度,从而可以识别GBM中的异质性。
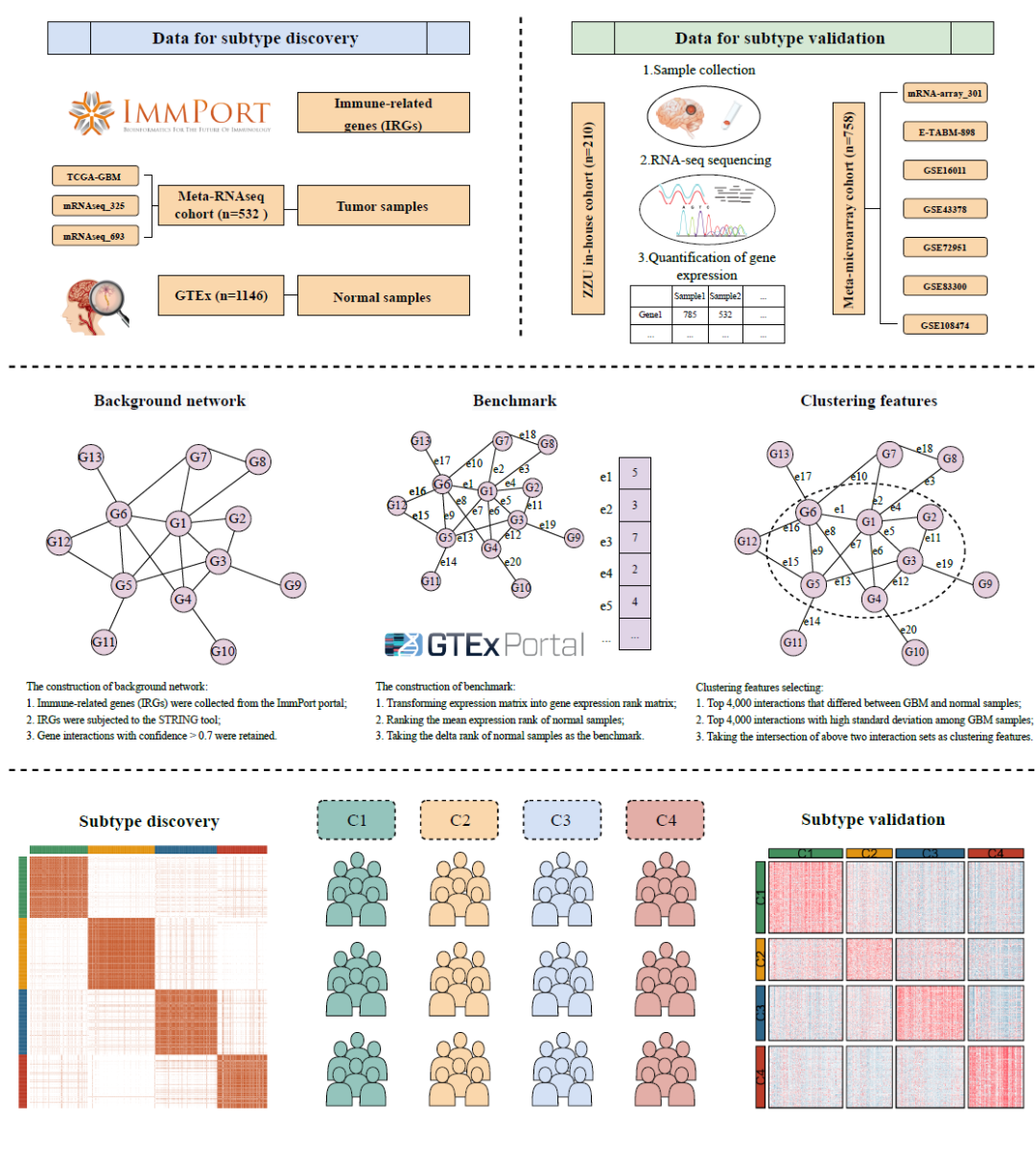
图1. 研究流程图
随后,在肿瘤样本中具有显著扰动和高异质性的1461个相互作用(由606个基因形成)被筛选出来用于共识聚类(表S3)。基于相互作用扰动矩阵,将Meta-RNAseq队列中的532个GBM样本通过共识聚类分为k(2~9)个组。共识矩阵、累积分布函数(CDF)曲线和差值面积图表明最佳聚类数为4个(图2A和图S2C、D),包括C1(131例样本,25%)、C2(150例样本,28%)、C3(139例样本,26%)和C4(112例样本,21%)。
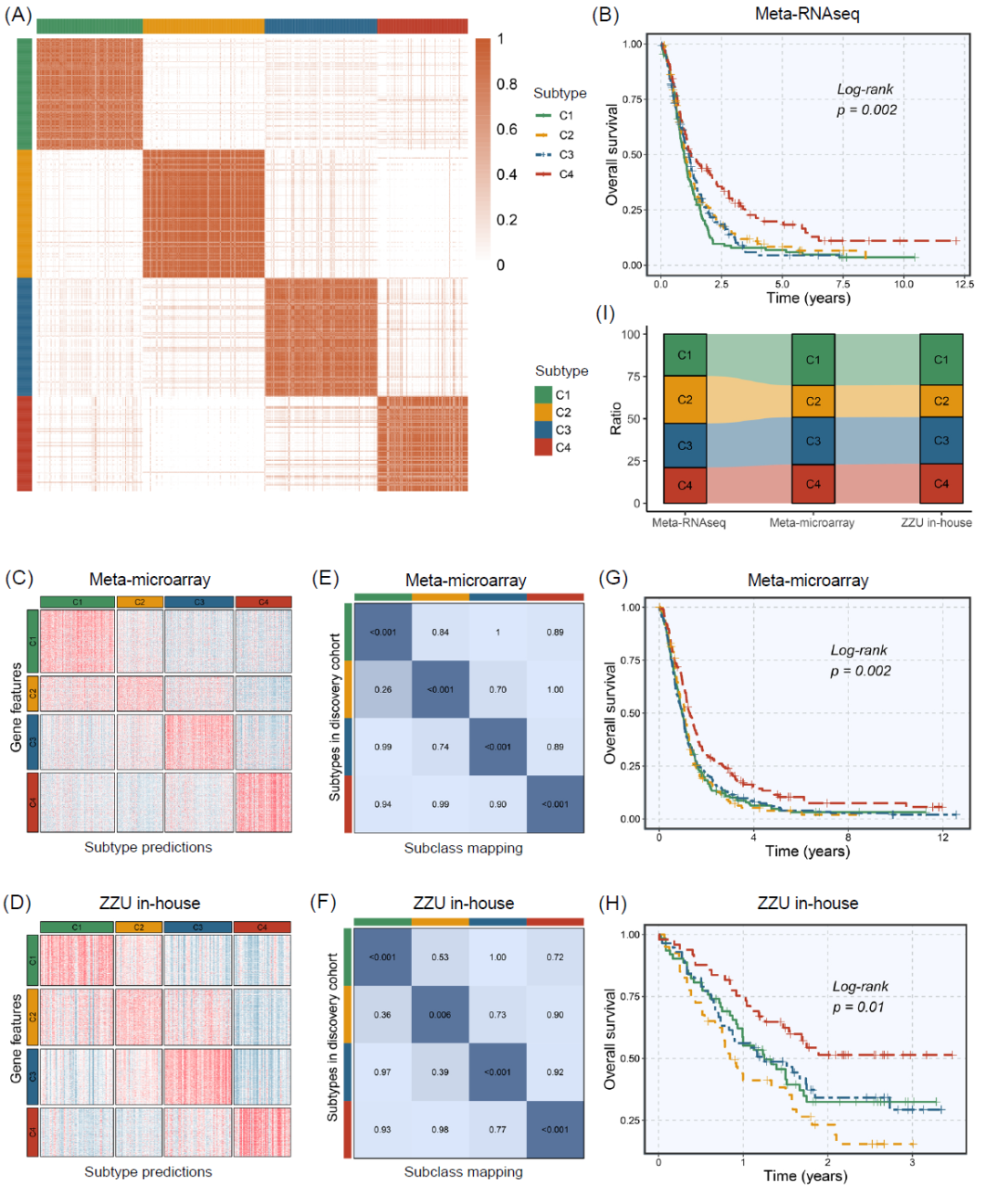
图2. 基于免疫基因相互作用扰动网络的四种亚型的识别和验证
(A)Meta-RNAseq队列的样本聚类热图。(B)Meta-RNAseq队列中四种亚型的总体生存(OS)的Kaplan-Meier及对数秩检验。(C,D)Meta-microarray队列(C)和郑州大学(ZZU)内部队列(D)的最近模板预测(NTP)热图。(E)Meta-microarray队列与发现队列之间的SubMap。(F)ZZU内部队列与发现队列之间的SubMap。(G,H) Meta-microarray队列(G)和ZZU内部队列(H)中四种亚型的OS的Kaplan-Meier和log-rank检验。(I)Meta-RNAseq队列、Meta-microarray队列和ZZU内部队列中C1、C2、C3和C4的比例。
Kaplan-Meier生存分析表明,四个亚型的预后存在显著差异,其中C4显示出最长的总生存期(p = 0.002,图2B)。众所周知,放疗和化疗是GBM患者的临床常规治疗方法,已被证明可以延长GBM的中位生存期。在这里,为了评估不同亚型对放疗和化疗的敏感性,我们根据不同治疗状态进行了亚组分析。对于未接受放疗的患者,四个亚型在预后上没有差异(p = 0.153,图S3A)。对于接受放疗的患者,四个亚型的预后存在显著差异(p = 0.006),其中C4显示出最佳预后(图S3B)。化疗结果也显示出类似的发现(图S3C-D)。总体而言,相对于其他亚型,C4患者可能对常规放疗和化疗更敏感,这也解释了其良好的预后。
在外部队列和内部队列中进行亚型验证
为了验证我们的分型在跨平台队列中的可靠性和重复性,本研究从公共微阵列数据集中检索了758个符合条件的GBM样本,并对我们医院的210个GBM样本进行了RNA测序。首先,通过差异表达分析识别了四个亚型的1200个标志基因(表S4),进而在外部Meta-microarray队列(图2C)和内部RNA-seq队列(图2D)中识别出发现队列中确定的四个亚型。亚类映射(SubMap)分析进一步证实了验证数据集中的四个亚型与发现队列中相应亚型具有类似的转录特征(图2E,F)。此外,与训练集一致,C4在验证队列中也表现出最佳预后(Meta-microarray:p = 0.002;ZZU内部:p = 0.01)(图2G,H)。除了类似的转录组和临床特征外,四个亚型在不同队列中的比例也相当(图2I),这进一步说明了四种亚型在跨平台队列中的可靠性。总体而言,不同测序技术和大规模数据的验证工作验证了我们分类方法的稳健性和普适性。
四种亚型潜在的生物学特性
为了进一步探索不同亚型的潜在生物学特点,我们进行了基于过表达代表性分析(ORA)和基因集变异分析(GSVA)的功能富集。具体而言,C1具有高免疫活性和低肿瘤纯度(图3A、E、F)。C2表现出肿瘤浸润性表型和中等免疫活性(图3B、E)。C3在细胞周期、E2F靶标和G2M检查点等增殖相关通路上富集(图3C、E)。此外,高肿瘤纯度的肿瘤在C3中富集,表明C3肿瘤的增殖特性(图3F、G)。C4与突触形成和信号传递相关,包括化学突触传递、跨突触信号传递和通道活性(图3D)。此外,突触相关基因的表达在C4中显著上调,表明C4中存在突触形成(图S4)。为了将我们的分类法与以前的分子亚型进行比较,我们分析了经典亚型(CL)、间质亚型(MES)和原神经亚型(PN)在四个亚型中的分布情况。具体而言,C1表现出较高的MES亚型组成比例,其次是C2和C3,而C4则不包含MES亚型。相反,当涉及CL亚型时,C4显示出最高的比例,其次是C3、C2和C1(图S5)。总的来说,我们将C1定义为免疫浸润型GBM,C2定义为侵袭性GBM,C3定义为增殖型GBM,C4定义为突触形成型GBM。
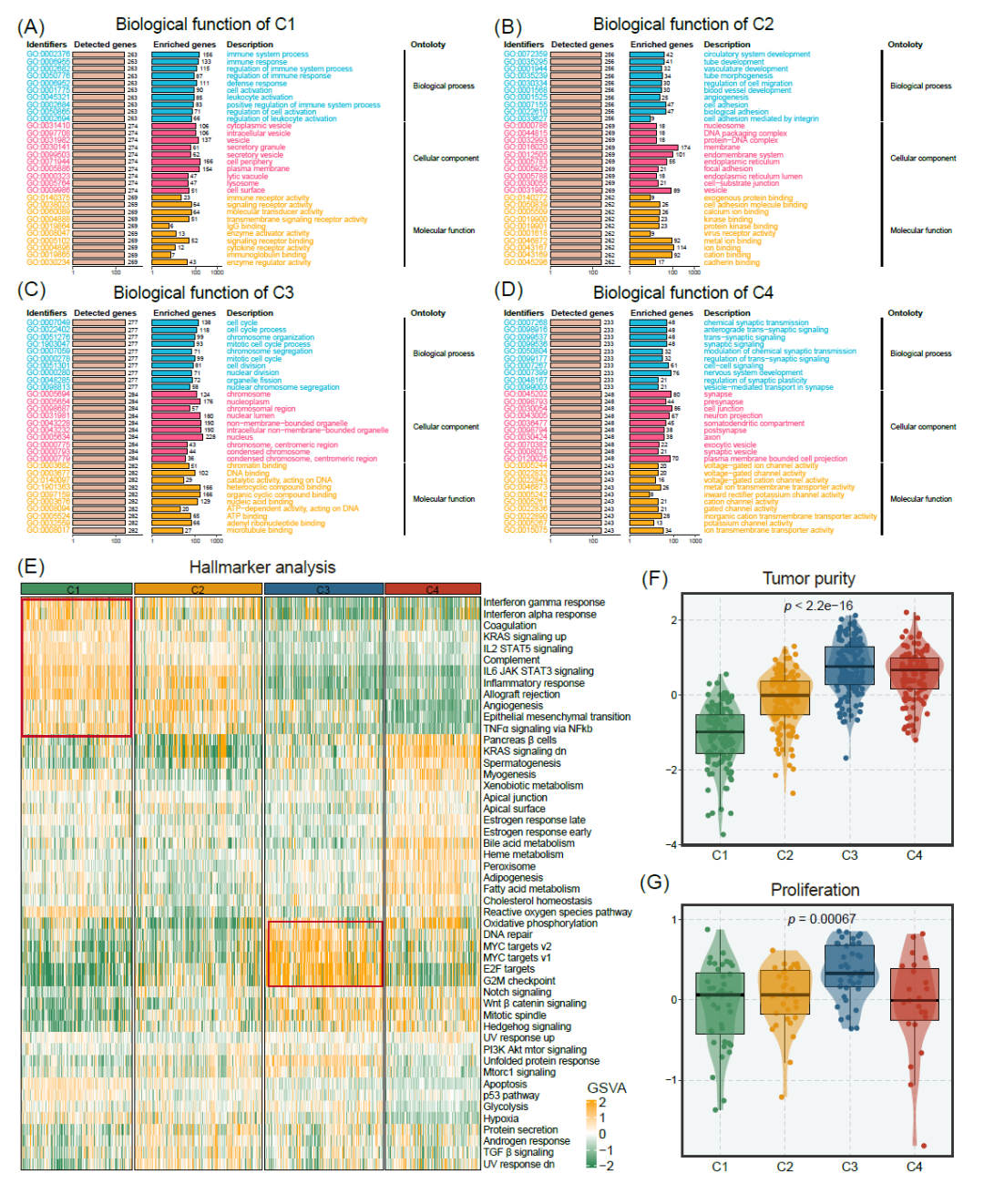
图3. 四种亚型的生物学功能和免疫浸润
(A-D)Meta-RNAseq队列中C1(A)、C2(B)、C3(C)和C4(D)的富集分析(GO)。(E) Meta-RNAseq队列中 "Hallmarker "基因组的GSVA分析,富集水平用z值表示。(F,G) Meta-RNAseq队列中肿瘤纯度(F)(用ESTIMATE算法测量)和四个亚型的增殖评分(G)。
四种亚型的代谢差异
为了探索四种亚型的代谢特征,我们通过GSVA分析了九个与物质相关的通路的富集情况(图S6)。值得注意的是,C1富集在外源生物降解代谢,表明存在升高的药物代谢,这可能导致药物耐药性。C2具有较高水平的葡萄糖代谢,尤其是糖酵解。高度的糖酵解活性可以通过乳酸发酵代谢大量的葡萄糖,即使在有氧条件下,这可能导致酸性环境,从而促进肿瘤血管生成。C3表现出较高的核苷酸代谢激活水平,这与其高增殖水平相一致。C4在氨基酸和脂质代谢通路富集,这有助于突触形成和信号传递。总之,这些发现展示了四种亚型中代谢相关通路富集的独特性,为我们的分型提供了更高的分辨率。
四种亚型的免疫景观和免疫治疗潜力
四种亚型在肿瘤微环境方面也具有显著差异。C1具有较高的免疫和基质评分(通过ESTIMATE算法测量),这表明C1中肿瘤纯度较低,微环境成分丰富(图4A和图S7A)。为了进一步量化四种亚型中的免疫浸润程度,我们通过单样本基因集富集分析(ssGSEA)对29个免疫标志物进行了分析(图S7B)。C1显示出最高的整体评分,其次是C2,而C3和C4则显示了较少的免疫浸润。抗原处理和呈递机制评分(APS)和MHC分子被用于描述肿瘤中的抗原处理和呈递能力,在C1中显示出更高水平(图4B、C)。此外,C1还显示出更多的免疫细胞浸润(通过ssGSEA估计),如CD8+ T细胞、CD4+ T细胞和自然杀伤细胞(图4D、E)。然而,CIBERSORT结果显示C1中的主要免疫细胞比例是M2巨噬细胞(图S7C)。我们进一步分析了四种亚型与免疫细胞以及肿瘤免疫循环之间的相关性。C1与大多数免疫细胞和肿瘤免疫循环呈正相关关系(图4E和图S8)。有趣的是,C1表现出更高的免疫检查点、共刺激和共抑制分子的表达(图4D和图S7D-E)。因此,C1肿瘤可能从免疫治疗中获益。
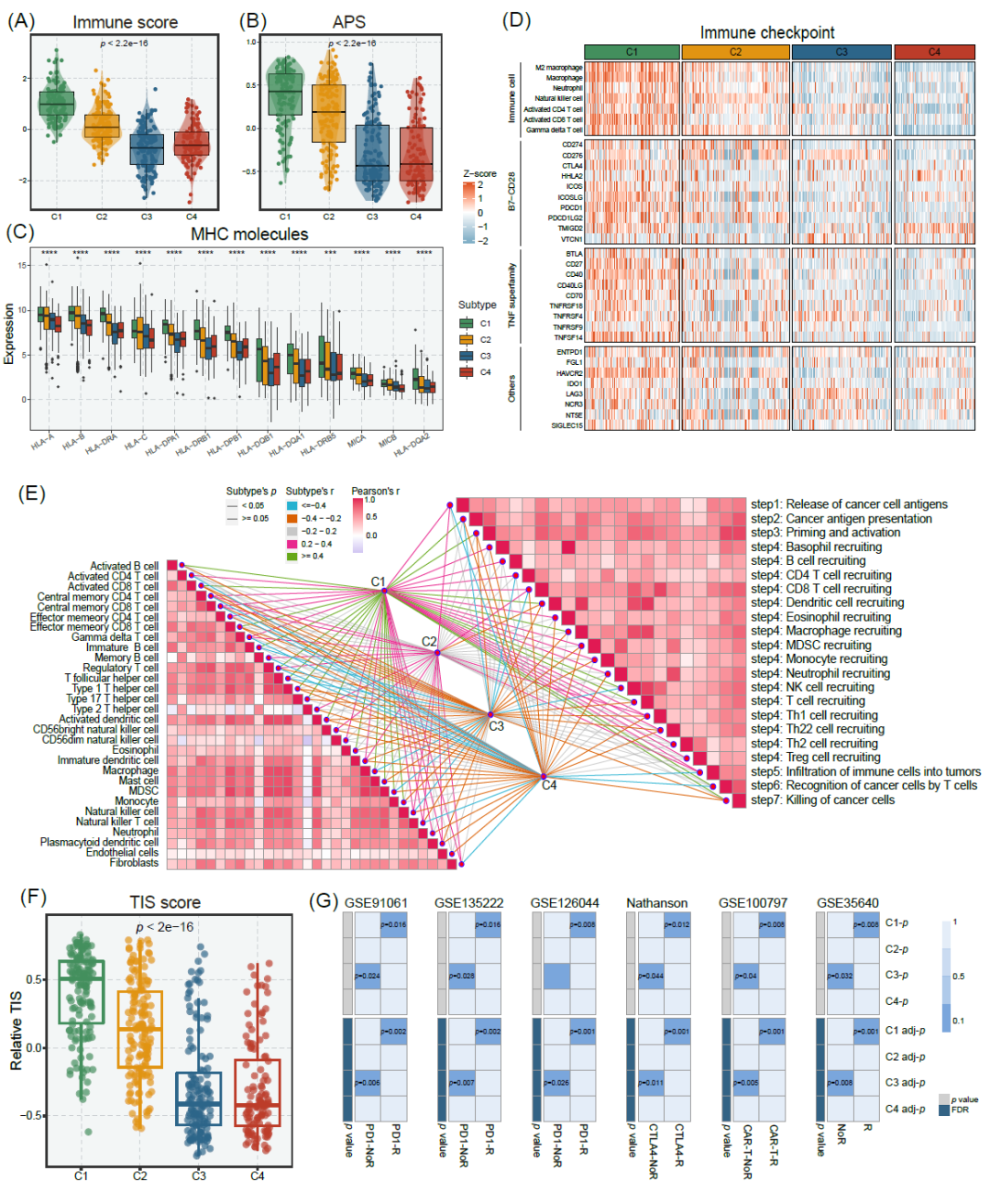
图4. 四种亚型的免疫浸润和免疫治疗预测
(A)四种亚型的免疫评分。(B)四种亚型的相对抗原处理和呈现机制评分(APS)。(C)四种亚型的MHC分子表达。(D)所有样本中7种免疫细胞群和27种免疫检查点分子的热图。免疫细胞的相对浸润丰度和免疫检查点的表达用z值表示。(E)四种亚型与肿瘤免疫循环和免疫细胞表达的相关性。(F)四种亚型的肿瘤炎症特征(TIS)评分。(G)四个亚型和六个免疫治疗队列(GSE91061、GSE135222、GSE126044、Nathanson、GSE100797和GSE35640)的SubMap分析,所有队列都有详细的免疫治疗信息。在SubMap分析中,p值越小意味着配对表达谱的相似性越高。*** p < 0.001, **** p < 0.0001。
为了证实我们的推测,我们应用了两种生物信息学算法,包括肿瘤炎症特征(TIS)评分和SubMap,以评估四种亚型免疫治疗的疗效。TIS评分用于测量肿瘤内现有的免疫抑制反应。一项关于前瞻性免疫治疗队列的临床研究显示,TIS评分高的患者在免疫治疗中获益更多。本研究中,C1具有最高的TIS评分,而C3的评分最低(图4F)。此外,我们还进行了SubMap分析,以阐明四个亚型与对免疫治疗的不同反应患者之间的表达模式的相似性。正如预期的那样,C1与所有免疫治疗队列中的免疫治疗应答者具有相似转录特征(图4G)。相反,C3显示出与免疫治疗不应答者类似的基因表达模式(图4G)。之前的结果中,C3具有高肿瘤纯度和匮乏的免疫浸润,这表明C3以免疫荒漠表型和免疫治疗的不足免疫储备为特征。我们还根据对合并的免疫治疗队列进行了NTP验证,并比较了四个亚型对免疫治疗的反应率。结果显示在图S9中,与先前的研究结果一致。值得注意的是,尽管这些免疫治疗队列并非来自GBM患者,但仍可以为我们的亚型探索提供参考价值,这在先前的文献中也有相似应用。
上述多个生物信息学方法的结果表明C1亚型可能从免疫治疗中获益,而C3亚型对免疫治疗不应答。为了进一步证实这些发现,我们对四个亚型进行了PD-L1的免疫组化染色。每个亚型随机选择了代表性样本进行PD-L1染色,并对PD-L1表达进行统计分析,结果显示C1的PD-L1表达水平明显升高,而C3的水平相对较低(图S10A,B)。
四种亚型具有不同的基因组特征
GBM具有显著的基因组不稳定性和分子异质性。在本研究中,TP53、ATRX和PIK3CA突变在C3中普遍存在(图5A)。NF1突变可导致神经纤维瘤蛋白缺失和随后的RAS活性升高。它被认为是胶质瘤耐药的生物标志物,在C4中的发生率很低(图5A)。我们还比较了可能降低NF1表达的17q11.2缺失的比例(图S7F)。在四个亚型中,C2出现25%的缺失,其次是C1(21%)、C3(11%)和C4(5%)。此外,在4个亚型中还探索了O6-甲基鸟嘌呤甲基转移酶(MGMT)启动子的状态。MGMT甲基化状态可提高对化疗药物的敏感性。尽管四种亚型的MGMT甲基化状态在统计学上没有差异,但MGMT启动子甲基化在C1-4中逐渐增加,其中C4型的MGMT启动子甲基化程度最高(图S11)。此外,我们还比较了Meta-RNAseq和ZZU内部队列中IDH和TERT启动子(TERTp)的突变频率(图5B-D)。C4的IDH突变率最高,与C4的良好预后相对应(图5B, C)。此外,作为临床预后不良的独立指标,TERT启动子突变在C2中富集(图5D)。此外,我们观察到G-CIMP状态和chr1p19q缺失状态在C4中尤为明显,这两种状态都是预后的有利因素,进一步验证了C4的最佳临床结局(图5E,F)。总之,这些结果表明四个亚型具有不同的基因组特征,可能驱动不同的生物学特性。
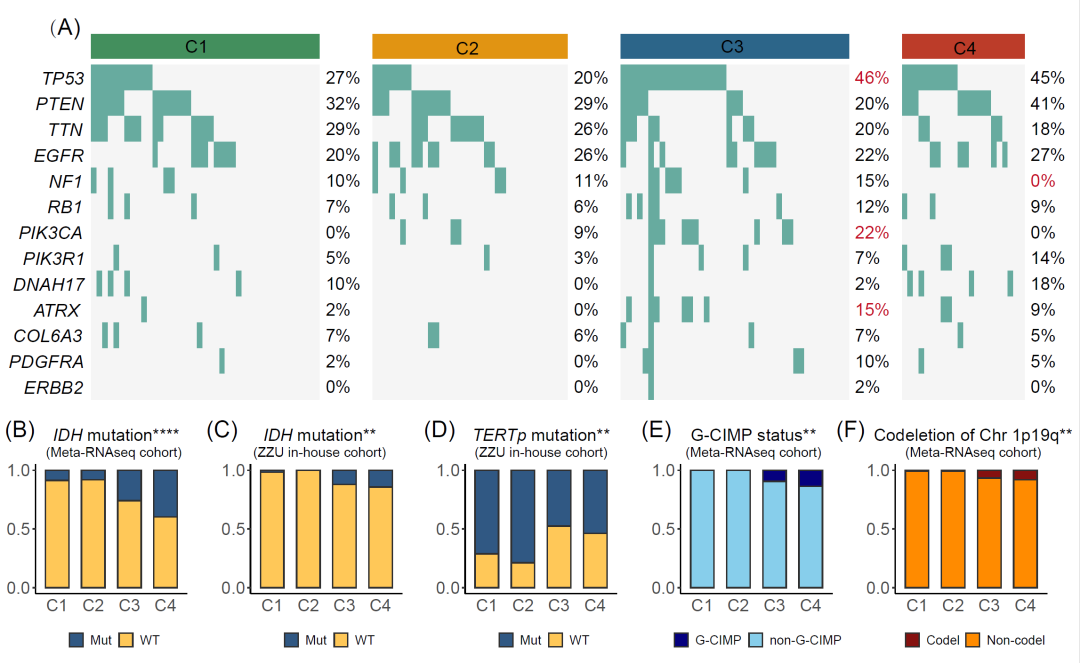
图5. 四种亚型的基因组特征图谱
(A)四种亚型中基因突变的瀑布图。(B)Meta-RNAseq队列中IDH的突变。(C,D)ZZU内部队列中IDH(C)和TERT启动子(TERTp)(D)的突变。(E,F)四个亚型中G-CIMP状态(E)和chr 1p19q缺失(F)的比较。** p < 0.01, *** p < 0.001, **** p < 0.0001。
四种亚型的潜在治疗药物
上述结果展示了四种具有不同特征的亚型,为基于亚型的靶向治疗提供了方向。为了更好地推进临床治疗,本研究引入了药物预测,以确定四种亚型的潜在治疗药物(图S12)。
研究发现,阿西替尼、马西替尼、OSI-930和帕唑帕尼这四种靶向RTK信号转导的成分在C2中具有优先作用。在RTK信号转导中,PDGF/RTK和VEGF/RTK与肿瘤的生长过程密切相关,特别是在促进肿瘤血管生成方面,这与C2的侵袭性特点相一致。此外,5种靶向增殖的药物可能对C3亚型有效,包括抑制染色质组蛋白乙酰化的伏立诺他,抑制DNA复制的顺铂、阿糖胞苷和替莫唑胺,以及靶向PI3K信号转导的MK-2206,这也与C3的增殖特性相吻合。在C1中也发现了几种具有抗癌活性的药物,如细胞凋亡调节、细胞周期、JNK和p38信号转导以及细胞骨架。结合C4对化疗更敏感的发现,我们推测C4可能对多种化疗药物有更好的反应性。然而,C1中仅含有一种仍在临床开发中的药物。这可能是由于C1中的肿瘤纯度较低,影响了化疗药物的有效性。如前所述,C1表现出对免疫治疗的敏感性,这可能成为C1最有利的治疗方法。总之,我们发现了针对四种亚型的特异性干预措施,这也与它们各自的生物学特征相吻合。
讨 论
GBM中的癌细胞产生了一种促血管生成和炎症的微环境,这种微环境招募了免疫细胞和分子渗透到肿瘤组织中,导致了GBM中错综复杂的免疫网络。新的证据表明,由肿瘤微环境中多种免疫成分组成的免疫网络可以描述肿瘤的免疫表型并影响GBM患者的临床预后。由于免疫网络的重要作用及其在GBM中的多样化存在,本研究基于免疫相关基因进行了聚类分析,这有助于理解GBM的异质性并促进个体化治疗。
基因表达谱存在变异,在不同时间点或不同条件下评估时可能表现出差异性,这导致基于表达数据构建的亚型具有不稳定性。为了解决表达谱分析的快照效应,我们利用相对稳定的基因相互作用扰动网络来构建分子亚型。此外,与单基因特征相比,基于网络的特征表现出较高的弹性和有效性,已被广泛接受用于分析高通量数据。然而,既往文章中的方法大多只利用网络中的基因集。与以往的方法不同,该研究中基于网络的方法不仅关注基因组中的分子,还关注它们之间的相互作用,这反映了每个分子不是独立发挥功能的事实。我们基于免疫相关的相互作用扰动网络,确定了具有不同生物学特征的四种亚型。该分类法在跨平台数据集中被证明是稳定和可重复的,其显示了类似的转录组特征、临床结果和可比比例。此外,四种亚型还展现了不同的生物学特征和分子可解释性,这可能对临床分层管理和基于亚型的特定干预措施有所启示。
C1为免疫浸润亚型,免疫浸润程度高,基质评分高,肿瘤纯度低,NF1表达缺乏,MES样亚型,预后差,对免疫治疗有潜在反应。高水平的APS和丰富的免疫细胞浸润倾向于激活C1的抗肿瘤免疫。肿瘤免疫循环是指在抗癌免疫反应中有效杀伤癌细胞的启动、进行和扩展的一系列步骤,C1和肿瘤免疫循环之间的正相关进一步验证了C1的免疫浸润表型。C1的高TIS评分意味着其从免疫治疗中获益的潜力,这一点通过SubMap分析和实验证据得到了进一步验证。然而,免疫检查点和共抑制分子表达水平升高的肿瘤也在C1中富集,这表明C1可能在刺激免疫激活后通过过度表达免疫抑制分子来逃避免疫清除。此外,在C1中发现了NF1的缺乏,这与M2巨噬细胞的高浸润是一致的。GBM中的M2巨噬细胞亚型代表肿瘤支持性巨噬细胞,具有促进肿瘤发展的能力,这可能是C1预后不良的部分原因。肿瘤纯度低、NF1表达缺乏和M2巨噬细胞浸润是MES亚型的特征,预后较差。分子亚型的比较揭示了C1的MES样特征,与其不良预后相吻合。总之,尽管C1具有预后不良的特点,但C1的特殊性为筛选可能从免疫治疗中获益的GBM患者提供了启示。
C2和C3均表现为恶性表型,临床预后不佳。C2为侵袭性亚型,具有中度免疫浸润,大量TERTp突变,对RTK信号抑制剂敏感。作为GBM常见的驱动基因,TERTp突变是临床预后不良的独立指标,可导致端粒酶表达水平升高和肿瘤细胞永生化。C2中大量的TERTp突变意味着恶性表型,并与其不良预后相一致。与侵袭性特征相一致,4种靶向RTK信号转导的成分阿西替尼、马西替尼、OSI-930和帕唑帕尼对C2亚型具有潜在的治疗作用。C3为增殖亚型,具有 "免疫荒漠 "的肿瘤免疫微环境,肿瘤纯度高,TP53、ATRX和PIK3CA突变,对PI3K信号抑制剂和DNA复制抑制剂敏感。与增殖的特殊性相一致,C3中高的PIK3CA突变可激活PI3K/AKT信号,促进癌细胞的增殖,这可以解释C3对PI3K信号抑制剂的高敏感性。ATRX突变导致的ATRX失活与TP53突变有关,这与C3中ATRX和TP53突变频率最高相吻合。此外,C3的免疫治疗耐药可能是由于免疫细胞和免疫分子尤其是PD-L1的浸润较少,不能充分启动抗肿瘤免疫导致的。
C4为突触形成型GBM,预后最好,表型为 "免疫冷",IDH突变多,NF1突变少,较高的chr 1p19q缺失、MGMT启动子甲基化和G-CIMP水平,对放化疗具有潜在敏感性。尽管C4表现出“免疫冷”型,但其在四种亚型中预后最好。事实证明,高IDH突变与白细胞趋化性和肿瘤相关免疫细胞减少有关,从而导致生存时间延长,这与C4的特性相符。高MGMT启动子甲基化可能导致DNA烷基化修复效率低下,这与C4对化疗的敏感性有关。类似地,M2巨噬细胞是导致GBM放疗耐药的原因,而C4中M2巨噬细胞的低浸润可能是其对放疗敏感的原因。有趣的是,C4在突触形成和信号转导方面表现出明显的富集,提示C4肿瘤细胞可能与神经元有一定的联系。Venkatesh等研究发现,肿瘤细胞可以通过神经回路中的神经元-胶质瘤突触与神经元进行电化学通讯,神经元活动诱导的胶质瘤膜去极化可以引起胶质瘤增殖,而通过药物或基因阻断电化学信号传导可以抑制胶质瘤异种移植的生长,延长存活时间。因此,通过干扰神经元兴奋性的调节来抑制肿瘤生长可能是针对C4肿瘤的一种新的潜在治疗方法。
尽管我们的分型很有前景,但我们的研究也存在一些局限性。首先,本研究中的所有样本都是回顾性的,应该在前瞻性数据中进一步验证这种分类方法。其次,四种亚型的生物学特征需要通过实验来验证。第三,这些用于免疫治疗预测的免疫治疗队列并非来自GBM患者,临床GBM队列的免疫治疗信息需要进一步验证。第四,背景网络是通过STRING工具构建的,只反映了蛋白质与蛋白质之间的相互作用。虽然蛋白质之间的相互作用网络可以在很大程度上代表基因与基因之间的相互作用,但它可能会干扰目前的结果。最后,由于公共数据库的限制,Meta-RNAseq和Meta-microarray队列中没有2021年WHO对CNS肿瘤的分类来定义的GBM信息。
结 论
我们采用了一种基于网络的方法来构建免疫相关的相互作用扰动网络,进而确定了具有不同生物学特性和临床结局的四种亚型。该研究提高了我们对GBM异质性的认识,有利于临床对GBM患者进行分层管理和精准治疗。
方 法
数据来源和处理
GBM RNA-seq数据。RNA-seq数据来自The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) 和Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn/)。共有532个样本组成训练集,包括三个数据集, TCGA-GBM(n = 144),mRNAseq_325(n = 139)和mRNAseq_693(n = 249)(表S5)。
GBM芯片数据。微阵列数据收集自CGGA、ArrayExpress(https://www.ebi.ac.uk/biostudies/arrayexpress/)和Gene Expression Omnibus(GEO,https://www.ncbi.nlm.nih.gov/geo/)。共有758个样本组成外部验证队列,包括7个数据集,其中mRNA-array_301(n = 124)来自CGGA,E-TABM-898(n = 56)来自ArrayExpress,GSE16011(n = 165)、GSE43378(n = 32)、GSE72951(n = 112)、GSE83300(n = 50)和GSE108474(n = 219)来自GEO(表S5)。
正常脑组织RNA-seq数据。1146份正常脑样本的表达谱来自GTEx Analysis V8,该数据下载自Genotype-Tissue Expression (GTEx, https://gtexportal.org/)数据库,其中包含正常组织的RNA测序数据。
ZZU内部队列和RNA测序。从在郑州大学第一附属医院接受手术治疗的患者中收集了210份GBM样本,所有患者均签署了书面知情同意书。所有纳入的GBM组织均经郑州大学第一附属医院伦理委员会批准(编号:2019-KY-176)。样本符合筛选标准:(1)组织病理学鉴定为GBM;(2)患者未接受新辅助化疗或放疗;(3)患者临床资料完整。根据之前的研究,对从GBM组织中提取的RNA进行定性和定量检查。采用NEBext® UltraTM RNA Library Prep Kit for Illumina®(NEB,美国)制备测序文库。使用TruSeq PE Cluster Kit v3-cBot-HS(Illumina)生成簇后,在Illumina Hiseq平台上对文库制备进行测序。对原始数据进行质控后得到的高质量清洁数据(clean reads)用于下游分析。清洁读数通过从基因组网站下载的参考基因组和基因模型注释文件映射到参考基因组。详细方法见Supping Information。ZZU内部队列的临床数据见表S6。
免疫治疗队列。我们的研究共纳入了6个免疫治疗队列,分别是GSE91061、GSE135222、GSE126044、GSE100797、GSE35640和Nathanson数据集。这些队列的基因表达数据和免疫治疗注释均可公开获取。根据实体瘤应答评价标准(RECIST,v1.1),免疫治疗应答者定义为完全应答(CR)或部分应答(PR),免疫治疗无应答者定义为疾病稳定(SD)或疾病进展(PD)。
TCGA-GBM的多组学数据。体细胞突变数据从TCGA网站下载。使用TCGA VarScan2管道获取体细胞变异信息。
数据处理。
(1)将TCGA、CGGA、ZZU和GTEx的RNA-seq数据转换为每百万千碱基转录本(TPM),并进一步进行log2转换。ZZU内部队列的基因表达矩阵如表S7所示。
(2)GSE16011、GSE43378、GSE108474和E-TABM-898等4个芯片数据集来自同一芯片平台(Affymetrix Human Genome U133 Plus 2.0 Array)。原始CEL文件通过affy软件包中的鲁棒多阵列平均(RMA)方法进行归一化处理。Illumina(GSE72951)和Agilent(mRNA-array_301,GSE83300)的微阵列直接下载。
(3)应用sva软件包中的ComBat算法去除不同技术平台间的批次效应。总之,亚型发现在合并的RNA-seq发现队列(Meta-RNAseq队列)中进行,并在合并的微阵列队列(Meta-microarray队列)、内部RNA-seq队列(ZZU内部队列)和合并的免疫治疗队列中得到验证(图1)。
基于免疫相关相互作用扰动矩阵的亚型发现
为了基于免疫网络的相互作用扰动解码肿瘤的异质性,我们构建了免疫相关的相互作用扰动矩阵,并进一步分析如下:
(1)首先,从ImmPort门户网站(https://www.immport.org/)提取免疫相关基因(IRGs),并使用STRING工具(https://string-db.org/)进行分析。置信度大于0.7的基因互作被保留用于构建背景网络和进一步分析(图1)。
(2)如前所述,我们将每个基因的表达量在每个样本的所有基因中进行了排序。因此,将基因表达矩阵转化为基因表达秩矩阵。根据基因相互作用的背景网络,通过减去基因相互作用连接的基因对中两个基因的秩,得到delta秩矩阵(行代表基因相互作用;列代表样本)。例如,我们假设和RA,S 和 RB,S分别代表样本S中基因A和基因B的表达秩,这两个基因通过相互作用E连接在一起。
(3)我们对所有正常组织的平均表达量进行排序,计算出delta秩向量作为基准。然后用每个样本的delta秩减去基准值来生成基因相互作用扰动矩阵(图1)。例如,我们假定ẟ ̅E代表相互作用E在基准中的秩次,ẟE,S代表相互作用E在样本S中的delta秩次,那么相互作用E在样本S中的扰动(ΔE,S)等于ẟE,S 减去 ẟ ̅E。本研究重点关注肿瘤组织的相互作用扰动矩阵,以解读GBM的异质性亚型。
(4)聚类特征的选择基于两个标准:能够区分正常组织和肿瘤组织以及在GBM中保持显著的异质性。如之前报道的,根据Wilcox检验的p值,选择了GBM和正常样本间差异最大的4000个相互作用,同时也选择了GBM样本间标准偏差(SD)最大的4000个相互作用(图1)。对上述两组相互作用取交集用于共识聚类(图1)。
(5)在发现队列中,通过ConsensusClusterPlus软件包进行共识聚类来解读潜在的分子亚型(图1)。聚类距离选择欧几里得,聚类算法选择Partitioning Around Medoid (PAM)。为确保聚类结果的稳健性,进行了1000次重复。通过累积分布函数(CDF)曲线确定最佳聚类数目。
亚型验证
最近模板预测(Nearest Template Prediction,NTP)是一种基于单个样本进行置信度评估的灵活的分类预测。为了评估从发现队列中产生的簇的可重复性,Meta-microarray队列和ZZU内部队列中的GBM患者根据特征基因使用NTP进行分类,特征基因由每个亚型的差异表达基因产生。FDR设定为0.2。SubMap是一种无监督的方法,它可以估计两组间共性的显著性,调整后的p < 0.05表示相似性显著。我们采用SubMap估计了两个验证队列和发现队列之间的亚型一致性。结果通过 "ComplexHeatmap "软件包显示。
统计分析
功能分析、免疫浸润评估、代谢分析、癌症-免疫周期、TME特征分析、药物预测、免疫组化(IHC)染色和免疫治疗反应评估的详细方法见Supping Information。所有数据处理、统计分析和绘图均在R 4.1.3软件中进行。Kaplan-Meier法和对数秩检验用于估计和比较不同亚型的总生存期(OS)。Wilcox检验或T检验用于比较两组间连续变量的差异,Kruskal Wallis检验用于比较三组或三组以上的数据。对分类变量的统计采用Chi-square检验或Fisher精确检验。FDR检验用于校正p值。通过Pearson相关分析确定相关性。所有统计p值均为双侧。P<0.05被认为具有统计学意义。
引文格式:
Zaoqu Liu, Yudi Xu, Yuhui Wang, Siyuan Weng, Hui Xu, Yuqing Ren, Chunguang Guo, Long Liu, Zhenyu Zhang, Xinwei Han. 2023. “Immune-related interaction perturbation networks unravel biological peculiars and clinical significance of glioblastoma.” iMeta e127. https://doi.org/10.1002/imt2.127
作者简介

刘灶渠(第一作者)
● 北京协和医学院博士在读,硕士毕业于郑州大学第一附属医院(导师:韩新巍教授)。
● 研究方向:生物多组学在肿瘤诊疗的应用。

徐雨迪(第一作者)
● 郑州大学第一附属医院硕士研究生在读。
● 研究方向:生物多组学在胶质瘤诊疗中的应用。

王宇慧(第一作者)
● 武汉大学医学硕士,从事分子遗传与细胞遗传实验室工作多年,擅长遗传病的临床数据分析及报告解读。
● 研究方向:肿瘤标志物及产前诊断。主持河南省医学科技攻关项目1项,以第一作者发表SCI论文7篇。

韩新巍(通讯作者)
● 二级教授,博士生导师,中原学者,郑州大学第一附属医院放射介入科主任,河南省介入放射学重点实验室主任。
● 从事影像诊断和介入放射学近30年,发明10余项介入新技术,开发10余个自主知识产权的介入新器械,攻克多项世界医学难题,是系列韩新巍式内支架的发明人。承担国家863项目1项,主持省部级科研攻关项目10余项。发表研究论文600余篇,其中SCI收录论文277篇,以通讯作者身份在European Heart Journal、Molecular Cancer、Circulation、Bioactive Materials和Nature Communacations等知名期刊发表SCI文章200余篇。担任Frontiers in Immunology,《介入放射学杂志》《影像诊断与介入放射学杂志》《中国介入影像与治疗学杂志》等杂志主编或编委。专业国际学术论文影响力中国排名前10。取得国家专利100余项,实现产品转化10余项。主编出版介入医学专著16部,其中英文专著《Airwaystenting in interventional radiology》,由全球最大科技出版社Springer(斯普林格)出版。获得中华医学科技奖三等奖,中国抗癌协会科技奖三等奖1项,军队科技进步奖,省部级科技进步奖一等奖2项,省部级科技进步奖二等奖4项,省部级科技进步奖三等奖4项,河南省医学科学技术普及奖一等奖1项。2021-2022入围国际前10万顶尖科学家。

张振宇(通讯作者)
● 博士,郑州大学第一附属医院神经外科副主任医师,胶质瘤亚专业副组长,河南省医学会神经外科分会青年委员会副主任委员,入选省级青年人才计划。
● 以(共同)通讯/第一作者发表SCI 30余篇,影响因子共计200余分;主持国家自然科学基金面上项目1项,国家自然科学基金青年基金1项,省级项目3项;担任EbioMedicine等杂志审稿专家。

刘龙(通讯作者)
● 西安交通大学第一附属医院博士在读,硕士毕业于郑州大学第一附属医院。
● 研究方向:肿瘤免疫、多组学与精准医学。发表SCI论文多篇,在 Nature Communications, EbioMedicine, Clinical and Translational Medicine 等知名杂志上共发表过 40 篇 SCI 论文。曾任Frontiers in Molecular Biosciences 杂志 Topic 客座副主编;任 BMC 系列、Lancet 系列和 Taylor & Francis 系列等多种杂志审稿专家。
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]