点击蓝字 关注我们
一种用于理解人类肠道微生物组在二型糖尿病中相关变化的神经网络框架

https://doi.org/10.1002/imt2.20

5.5
iMeta
RESEARCH ARTICLE
● 2022年5月5日,深圳先进技术研究院马迎飞、姜青山团队合作在iMeta在线发表题为“A neural network-based framework to understand the type 2 diabetes-related alteration of the human gut microbiome”的研究性文章。
● 该研究开发了一种结合神经网络和随机森林的框架,从宏基因组数据集鉴定出40个标记物种和90个标记基因,在预测中获得了更高的准确性,提供了一种新的思路来识别疾病相关生物标记物并分析它们在疾病发展中所扮演的角色。
● 第一作者:郭顺
● 通讯作者:马迎飞、姜青山(中国科学院深圳先进技术研究院)
([email protected]),([email protected])
● 合作作者:张皓然,楚云猛
摘 要
微生物标记物的识别有助于理解复杂的人类肠道微生物群在相关疾病的变化情况,因而受到广泛关注。在这里,我们开发了一种结合神经网络和随机森林的框架,从宏基因组数据集(185份健康样本和183份二型糖尿病样本)中分别鉴定出40个标记物种和90个标记基因。相比其他方法,神经网络模型根据这些标记物在预测中获得了更高的准确性。标记物的交互网络分析识别出了重要物种和功能模块。回归分析表明空腹血糖是人体肠道微生物组在二型糖尿病的相关改变中最重要的因素(P <0.05)。我们还观察到一些在健康组和对照组样本中变化很少的标记物种在二型糖尿病的不同阶段变化显著,这些表明它们在二型糖尿病相关的微生物组改变中的重要作用。我们的研究提供了一种新的思路来识别疾病相关生物标记物并分析它们在疾病发展中所扮演的角色。
关键词:人类肠道微生物群、神经网络、随机森林、二型糖尿病相关生物标记物
亮 点
● 一种结合神经网络和随机森林的框架,用于识别2型糖尿病相关生物标志物
● 构建生物标志物的定向相互作用网络分析相关微生物群落的在二型糖尿病相关变化中的潜在驱动因素
● 分析二型糖尿病发展过程中空腹血糖动态变化与生物标志物的协调变化
视频解读
Bilibili:https://www.bilibili.com/video/BV1nY4y1r7oD/
Youtube:https://youtu.be/ML48BEuWmmo
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
人体肠道微生物群是一个复杂的生态系统,包含10^14个微生物细胞,并被认为在人类健康和各种疾病中扮演着重要角色。随着下一代的测序技术到来,大量的微生物宏基因组测序数据从人类肠道样本中获得。元基因组研究提供了很好的机会,可以通过各种成熟的生物信息学工具和算法获得关于肠道微生物群如何与各种人类疾病相关的宝贵见解。在微生物组研究领域,一种常见的做法是依据疾病状态利用基于统计的策略(例如,Spearman的相关系数,Wilcoxon Rank-Sum测试等)来识别与肠道微生物群体相关的生物标志物,例如广泛应用的软件LEfSe。这些方法可以识别宏基因组特征,这些特征在病例组和对照组之间具有统计学上的显着差异。然而,这些基于统计的分析通常基于独立或线性假设,而肠道菌群的生态系统很复杂,很可能取决于许多微生物的非线性影响。为此,这些方法可能会忽略一些潜在的宏基因组生物标志物,这些标记物可能会导致人类肠道微生物组的疾病相关改变,但在样品中没有可检测到的显著的统计变化。例如,据报道,Veillonella parvula与二型糖尿病(T2D)有关,但是,在样品中的丰度几乎没有变化。
机器学习最近在生物学研究中引起了越来越多的关注。一些著名的算法包括支持向量机 (SVM)、随机森林 (RF)、隐马尔可夫模型 (HMM)、贝叶斯网络 (BN) 以及高斯网络,已应用于蛋白质结合的预测,微生物群落中的代谢功能,转录网络的表征,等等。由于机器学习可以生成模型并从大型数据集中找到预测模式,因此它将影响微生物组研究和其他生物学领域。然而,这些传统的机器学习算法存在一些局限性。例如,SVM 是一个线性模型,HMM 和 BN 依赖于一些基于概率的假设。为此,一些与 T2D 相关的物种,如 Coprobacillus catus 不会被这些方法鉴定,因为物种的丰度在样本之间变化不大(可能与疾病没有线性相关性)并且平均丰度非常低(在某些概率分布下将是异常值)。基于深度学习的方法通常使用具有多个隐藏层的神经网络 (NN),在最近的研究中显示出优秀的性能,因为它们可以识别给定复杂数据集中其他方法会忽略的新模式。最近,这些方法已应用于生物学领域,例如预测特殊基因/蛋白质功能、识别医学诊断、药物发现等。然而,在微生物组的相关领域应用该技术的一个限制是这些研究中可用样本的数量通常是有限的(例如,几百个样本)。至于深度学习模型,通常有很多层和神经元(决定模型中参数的大小),需要海量样本进行训练。除此之外,将 NN 模型应用于微生物组数据可能由于其“黑匣子”性质,仍然是一个关注点,也就是说,通常很难证明哪些输入特征在输出中起着决定性作用。
在这里,我们提出了一个结合 NN 和 RF 算法的框架,用于根据微生物丰度谱识别肠道微生物组与二型糖尿病相关的生物标志物。为了证明我们方法的实用性,我们使用了来自中国糖尿病患者和非糖尿病对照组的两个公开可用的独立的宏基因组数据集。根据已确定的标志物,我们首先证明空腹血糖(FBG)是与T2D 相关的人类肠道微生物组改变相关的最重要因素,并发现这些微生物标志物在病例和对照样本中变化不大,也可能发挥重要作用。我们的分析结果表明,这些标记物种而非单个物种的累积效应可能会驱动人类肠道微生物组的 T2D 相关改变。该研究为在微生物组研究中使用神经网络算法铺平了道路,并为深入了解微生物在人类疾病发展中的作用和评估有相关疾病风险的个体提供了潜在机会。
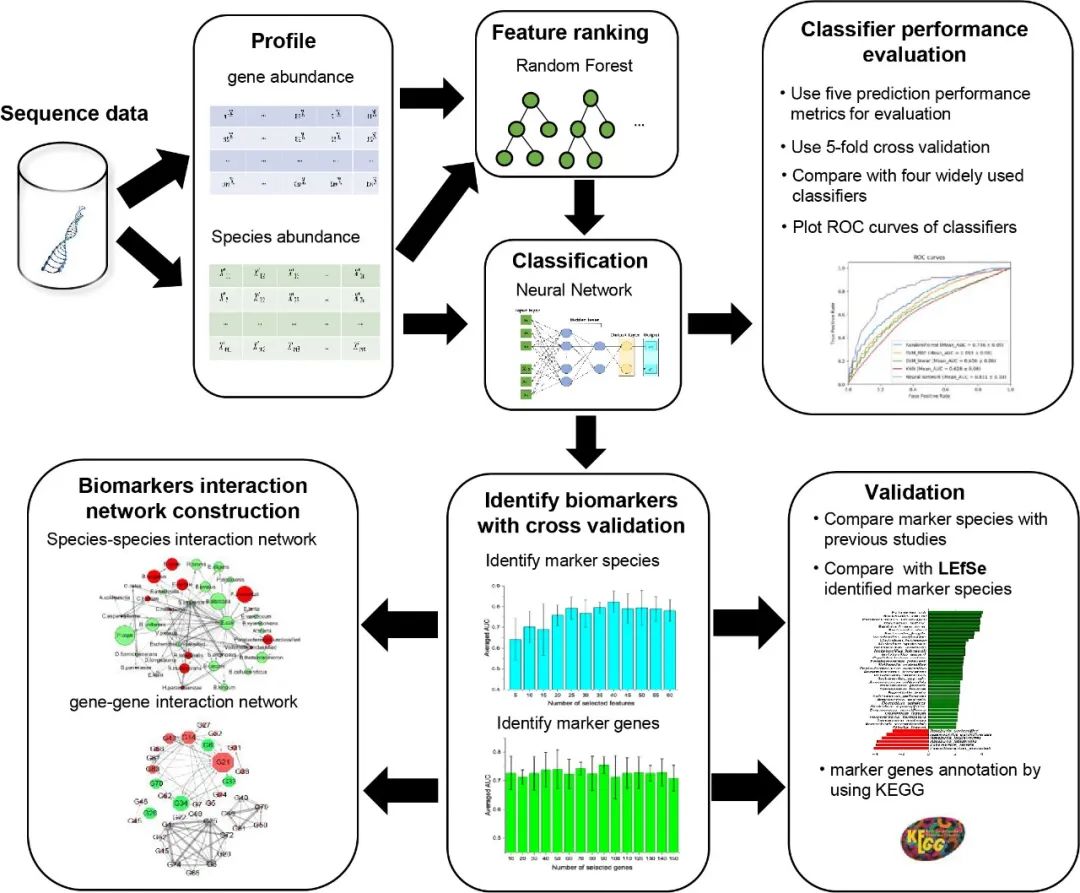
NN模型的性能优于其他方法
为了比较,我们评估了 NN 模型以及 SVM、SVM-RBF、RF 和 KNN 方法在对 D1 数据集的 T2D 相关样本进行分类时的预测性能。使用五折交叉验证(5 fold-CV)对微生物谱进行分析。接收者操作特征 (ROC) 曲线绘制在图 1 中,并且计算了每种方法的相应曲线下面积 (AUC) 指标。显然,与其他四种方法相比,NN 模型在所有指标上表现最好(AUC:~0.8,其他指标:~0.75)。这可能是由于NN模型的非线性拟合能力,它会提取更多的预测特征。至于其他四种方法,两个非线性分类器,即RF(AUC:0.736±0.05)和SVM-RBF(AUC:0.693±0.06)表现优于其余两种(SVM AUC:0.656±0.06,KNN AUC : 0.628 ± 0.06)。
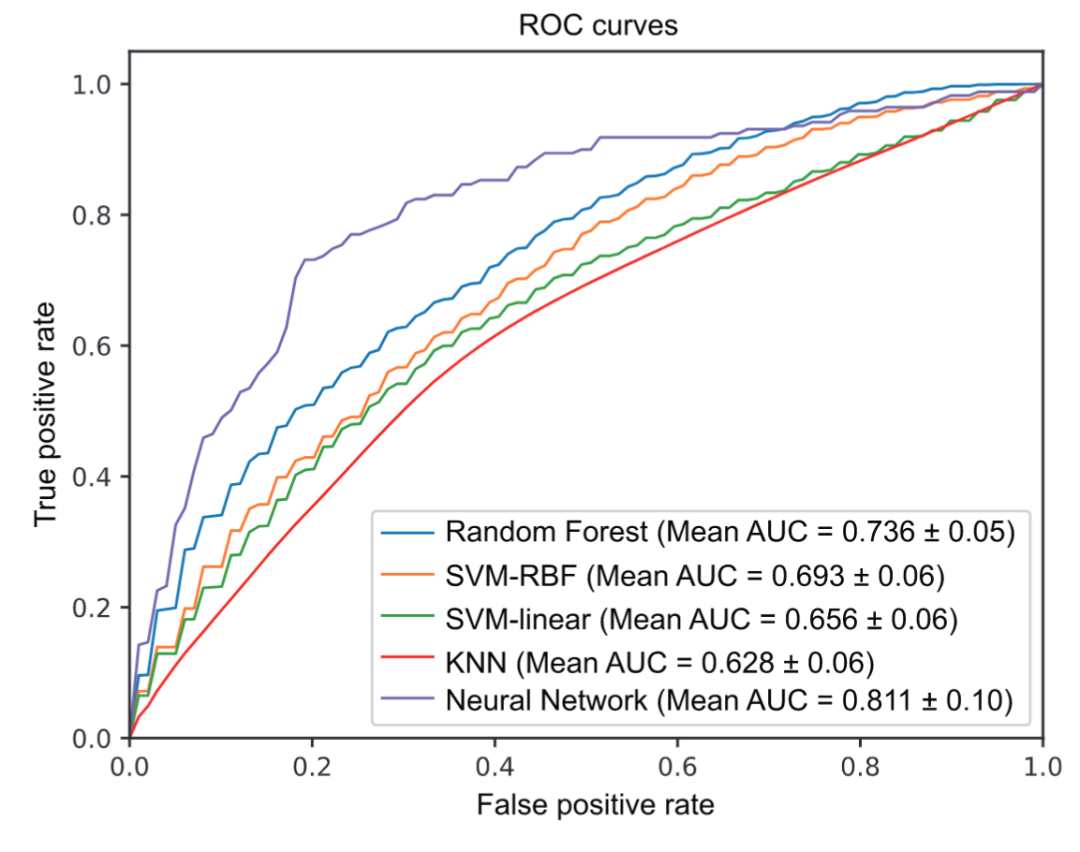
图 1. D1 数据集上 T2D 判定的交叉验证分析
通过五折交叉验证获得五个分类器(RF、SVM-linear、SVM-RBF、KNN、Neural Network)的接收器操作特征(ROC)曲线。AUC,曲线下面积;RBF,径向基函数;KNN,K-最近邻;RF,随机森林;SVM,支持向量机
识别的标记物种在人类肠道菌群的T2D相关变化中起决定性作用
我们进一步研究了所有肠道微生物群物种中的哪些物种,在使用 NN 模型对 T2D 相关样本进行分类中起决定性作用。由于 NN 模型的“黑匣子”性质,因此,我们使用广泛采用的特征选择方法RF, 根据其重要性分数对所有在 D1 样本中的物种进行排名。然后,我们将前 k个(k = 5、10、15、20、...、60)物种分别放入到 NN 模型中,用于对样本进行分类。在对 T2D 相关样本进行分类时,根据 5-fold CV 的平均 AUC 评估使用不同数量物种的 NN 模型的预测性能。从图 2A 可以看出,NN 模型在前40个物种中达到了峰值平均 AUC 值(82.3 ± 5%)。该预测结果甚至比使用所有物种(n = 270)的预测结果略好(一个原因可能是所有这些物种中都有一些噪声,因此会影响预测性能),表明选择的标记物种可以用于在一定程度上描绘与 T2D 相关的肠道菌群改变。因此,我们将选定的前40个物种作为标记物种。
为了评估我们选择的标记物种,我们选择了广泛应用的软件LEfSe来识别标记物种,巧合的是,也产生了与T2D相关40个标记(|线性判别分析)分数|>2)。值得注意的是,这40种标记物种中的16个与我们NN模型识别出来的相同,而其他24个标记物种则不同。此外,我们还使用软件 ANCOM-II来进一步验证我们的结果。有趣的是,ANCOM-II识别19个物种(临界值:0.7),而19个物种中有17个包含在我们选择的生物标志物中。然后,我们在 D1 数据集上使用5-fold CV 比较了五个分类器使用上述不同标记物种的预测性能。结果,我们方法识别的标记物在所有分类器上的预测性能比用LEfSe识别的生物标记物的预测性能表现更好(AUC 改进 ∼1% - ∼5%),而在其中两个(即 KNN 和 SVM-RBF)分类器上使用ANCOM-II 生物标志物的预测性能更好。一个可能的原因可能是 LEfSe 的算法忽略了一些具有判别能力的重要生物标记物,这些标记物丰度在病例和对照样本中几乎没有变化。
此外,我们采用了独立的数据集D2+来评估使用40个标记物种的NN模型的预测能力。在此测试分析中,我们将数据集D1-用于训练,而数据集D2+用于测试,预测性能AUC为76.2%。为了表征NN模型在D1样品中鉴定出的40种标记物种的变化,我们比较了它们在疾病和对照样品中的相对丰度的分布(图2B)。基于使用Kruskal-Wallis检验获得的结果,我们可以观察到,只有16个标记物在疾病和对照组样本中的相对丰度具有显著变化(P <0.05)。然而,一些标记物虽然相对丰度在所有样本中几乎没有变化,但在先前的研究中被识别出来与T2D相关(例如,在先前的研究中Catus和Collinsella aerofaciens)。观察结果表明,在所有样品中丰度没有显着变化的标记物,也可能在与T2D相关的肠道微生物改变中起特殊作用。
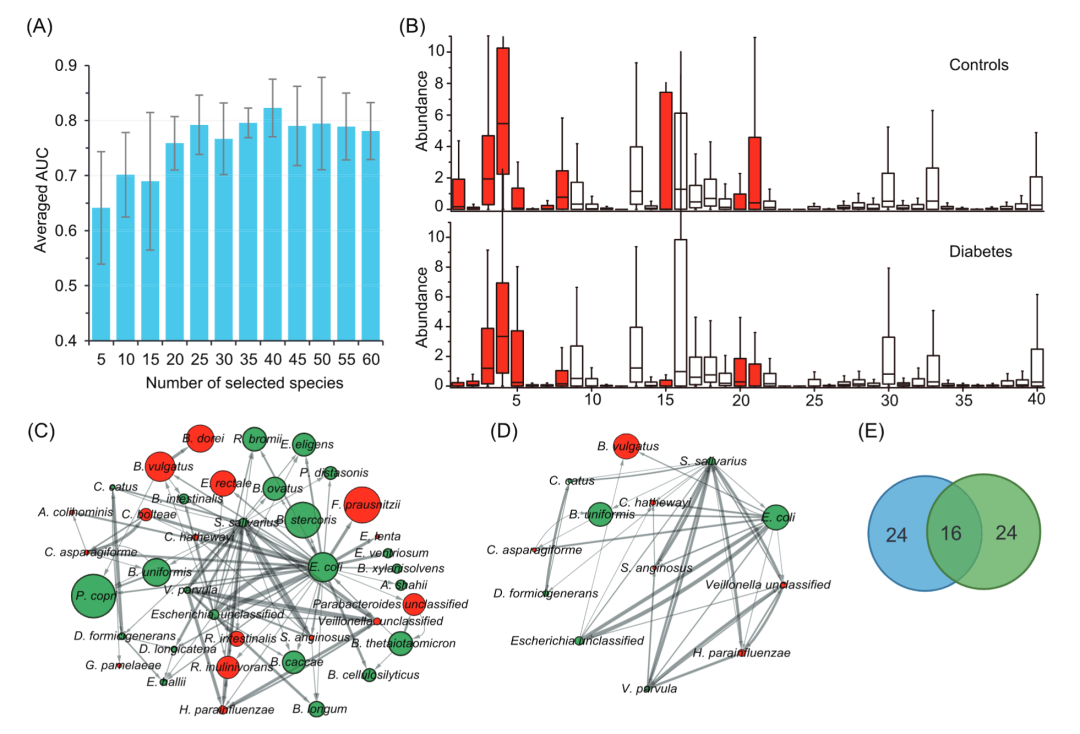
图2. 标记物种识别和交互网络构建
(A) 基于神经网络的分类器,在数据集D1上使用不同数量排序好的物种经过五折交叉验证获得的平均预测性能AUC。选择40个物种即可获得峰值,因此将这些物种确定为本研究的标志物种。(B) 糖尿病相关生态失调中标志物种的丰度分布。病例样本 (n = 183) 和对照样本 (n = 185) 之间的比较。使用 Kruskal-Wallis 检验,表明标为红色的物种的丰度在所有样品中有着显着变化(p < 0.05)。(C) 标记物种的相互作用网络由 GENIE3 生成,其中选择了标记物种之间最可靠的 100 个相互作用。在网络中,每个节点代表各自的标记物种;构建的网络是有向的,节点之间的箭头表示交互的方向,即节点A到B的箭头表示B的丰度主要受A的丰度影响。节点之间的边宽度与节点间连接的可靠性成正比。
相互作用网络识别出了与人类肠道微生物组改变相关的重要物种
为了进一步研究这些标记物种之间的关系,我们根据从数据集D1识别出的 标记物种的相对丰度构建了物种-物种相互作用网络。网络构建采用广泛应用的GENIE3,它通过考虑多个物种的非线性关系来计算相互作用。在本实验中,物种间所有可能的相互作用都根据 GENIE3 计算的可靠性得分进行排序,我们首先选择前 100个可靠性最高的相互作用关系来构建相互作用网络。如图 2C 所示,总共 40个标记物种中的 39个与其他物种至少有一种连接,其中13个种物种与至少 5个其他物种有连接(图 2D)。更有4个物种,包括 V.parvula、Streptococcus salivarius、Escherichia coli 和 Escherichia_unclassified 与其他物种有超过 10个连接。
此外,在病例和对照样品中,13个物种中的7个物种(绿色)的相对丰度变化不显著(图2D)。在这7个物种中,大肠杆菌和唾液链球菌的连接数分别为35和25,这表明这两个物种在人类肠道微生物组中与T2D相关变化中的具有相当的作用。大肠杆菌已被识别为糖尿病富集的物种,唾液链球菌可以发酵葡萄糖产生乳酸。在这13个(图2D)标记物中,将近70%(n = 9)是短链脂肪酸产生的细菌,并且先前的研究表明,这些类型的细菌在T2D中起重要作用。
另外,在13个标记物种中,除大肠杆菌,Bacteroides vulgatus和Bacteroides uniformis以外,其他物种在所有样品中的丰度都较低。这些基于选定标记物种的相互作用网络分析表明,丰度较低的物种也可能通过与其他物种相互作用在与T2D相关的变化中起到重要作用。因此,可以将具有五个以上连接的标记物种(n = 13)看作为与T2D相关的肠道菌群的核心。
识别的标记基因影响肠道微生物群与T2D相关的改变
为了鉴定与遗传和功能水平上与T2D相关的标记基因,我们还将方法应用于数据集D1上的功能基因谱。同样,我们使用RF方法对基因进行排序,然后使用五折交叉验证法评估了具有不同数量排序基因的NN模型的平均预测性能AUC。为此,我们选出90个基因作为标记基因。我们进一步使用90个标记基因(在D1-数据集中训练)在独立数据集D2+上测试了NN模型的预测性能,获得74.6%的AUC。以同样的方式,我们比较了这些标记基因的相对丰度在D1数据集所有样本中的变化情况,并观察到使用Kruskal -Wallis检验中的90个标记基因中的78个具有显着变化(P <0.05)(图3B)。
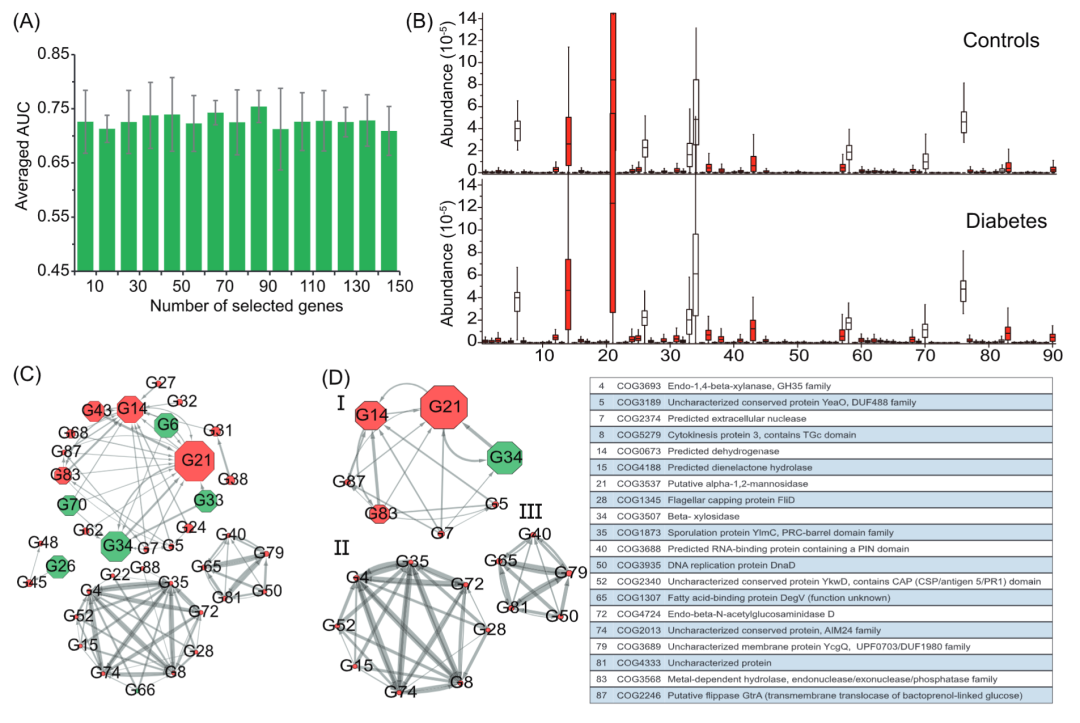
图 3 标记基因鉴定和交互网络构建
(A) 基于神经网络的分类器,在数据集D1上使用不同数量排序好的基因经过五折交叉验证获得的平均预测性能AUC。选择90个基因即可获得峰值,因此将这些基因确定为本研究的标志基因。(B) 糖尿病相关生态失调中标志基因的丰度分布。病例样本 (n = 183) 和对照样本 (n = 185) 之间的比较。使用 Kruskal-Wallis 检验,表明标为红色的基因的丰度在所有样品中有着显着变化(p < 0.05)。(C) 标记基因的相互作用网络由GENIE3构建,其中选择了标记基因之间最可靠的100个相互作用连接。节点间边的宽度与节点间连接的可靠性成正比。每个节点的大小与标记基因的平均丰度成正比。网络布局由 Cytoscape 软件使用圆形布局计算。每个节点中的数字是标记物种的 ID。红色的节点是在糖尿病样本或对照样本中显著富集的标记基因。(D) 连接数大于5的标记基因间的交互网络。
标记基因的相互作用网络识别与 T2D 相关的核心功能基因模块
为了表征标记基因之间的相互作用,我们同样使用 GENIE3(图 3C)构建了基因-基因相互作用网络。最终,在90个标记基因中有 37个基因与其他基因至少存在一个连接。如图 3C 所示,所构建基因间的相互作用网络可以划分为四个独立的组,每个组内的基因与该组内其他基因连接。少数基因(n = 20)与其他基因具有五个以上的连接(图3D)。我们利用BlastKOALA进行注释和基因并用KEGG映射来表征90个标记基因在功能模块或途径中的功能类别。结果,我们发现90个标记基因中的56.7%(n = 51)可以在直系同源组的数据库簇中注释(COGS)。90个基因中只有16个基因可以映射到KEGG数据库中的功能。根据标记基因在KEGG数据库中的功能注释,第I组基因中的4个(ID:34(CO G3507),70,70(COG2160),87(COG2246),68(COG1071),第II组基因中的2个基因(ID:48(COG 1211),26(COG0493)),第III组基因中的1个基因(ID:66(COG2971)被映射到碳水化合物代谢的途径,表明这些基因中可能参与碳水化合物相关代谢。第III组中的1个基因(ID:28(COG 1345)映射到鞭毛装配中。每组的基因与组内的其他基因相连,但不与不同组的基因相互作用。这表明不同组中的基因在碳水化合物代谢中的作用不同。这些基因的相互作用证实了人类肠道微生物组与T2D相关的改变之间的联系。
基于神经网络的回归分析揭示了与人类肠道微生物组在T2D相关变化中的最显著协变量
FBG、年龄、体重指数(BMI)和患者体重都可能是 T2D 的潜在致病因素。为了发现这些因素中最重要的因素,我们用选出的40个标记物种在数据集D1上进行了回归分析。基于五折交叉验证法,对于每个因素,通过NN模型计算样本的预测值。然后,绘制出这些预测值和相应的实际值散点图(图 4)。这些图表明,对于每个因素,基于标记物种的预测值与样本中相应的实际值存在一定程度的线性关系(图 4A-D),这也表明人类肠道微生物组与这些因素相关。特别是,我们观察到微生物组与 FBG (p < e-50) 的相关性比与其他因素(年龄,e-43;BMI,e-30;体重,e-32)更为显着。我们推断 FBG 可能是在 T2D 发展过程中驱动人类肠道微生物组T2D相关改变的主要因素。尽管有不少的研究工作报道了肠道微生物组与 FBG 之间的关系,但相比于传统的分类方法,我们通过非线性回归分析揭示了FBG 与人类肠道微生物组在T2D相关改变之间的较强相关性。
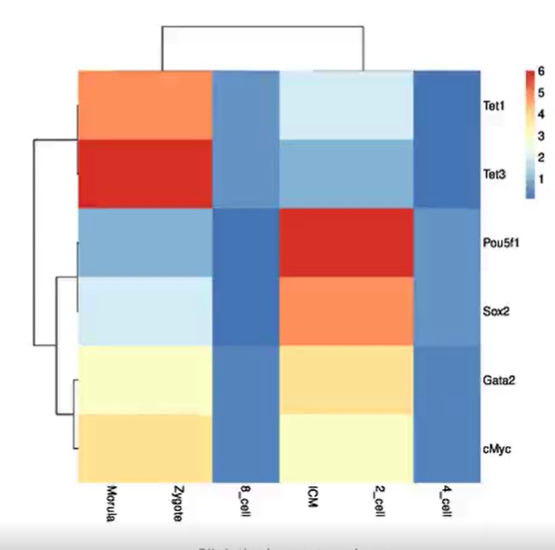
为了探索我们的标记物种在 T2D 发展过程中如何与 FBG 的动态变化共同变化,我们绘制了在FBG四个区间的标记物种平均相对丰度的热图(即 Q1:< 5.02;Q2:5.02-6.21 ; Q3: 6.21–8.8; Q4: > 8.8) (图 5)。可以观察到,这些标记物种的相对丰度在不同的区间间隔内变化很大(图 5)。基于Kruskal-Wallis 检验,有16个标记物种在病例和对照样本之间的相对丰度存在显着差异 (p < 0.05),其中有6个在健康对照样本中具有较高的相对丰度,另外10个在 T2D 样本中具有较高的相对丰度。这6个标记物(图 5,黑色)在 Q1 和 Q2 中的丰度显著高于在 Q3 和 Q4中的丰度。而在另外10个标记物种中,有6个在Q3 和 Q4 区间的丰度较高,有1个标记物种(Bacteroides dorei)在Q4 中的丰度较高,有1个标记物种(Eggerthella lenta)在Q3 中的丰度较高,而另外的两个标记物种(Veillonella. unclassified 和 Streptococcus anginosus)在Q1 和 Q3 中的丰度较高。这些标记物种很容易根据它们在T2D和健康样本中相对丰度的差异性,通过传统的统计的方法识别出来。另外24个标记物种在病例和对照样本之间相对丰度没有显着差异,这些标记物种表现出不同的模式。直观地说,其中一些标记物种(n = 3、Dorea longicatena、Prevotella copri 和 S. salivarius)在 Q1 中达到最高丰度,有两个(Bacteroides ovatus、Escherichia.unclassified)在Q2中达到最高丰度,有五个(V.parvula, Bacteroides xylanisolvens, Eubacterium eligens, R.bromii, Bifidobacterium longum)在 Q3 中到最高丰度;Q2 和 Q3 中达到最高丰度的有三个(Alistipes shahii、E.coli和 Ensete ventriosurn),Q2 和 Q4 中的达到最高丰度的有两个(Bacteroides thetaiotaomicron、Eubacterium hallii)。因此,这 24 种标记物种在以与 FBG 的动态变化相关的不同模式变化。这可能解释了交互网络分析检测到的两个重要的 T2D 相关标记物种(E. coli and S. salivarius)在病例和对照样本的丰度中几乎没有显著变化的原因。总之,我们的分析表明,这些标志物可能在 T2D 发展的不同阶段发挥不同的作用,然后这些标志物的累积效应驱动人类肠道微生物组的 T2D 相关改变。
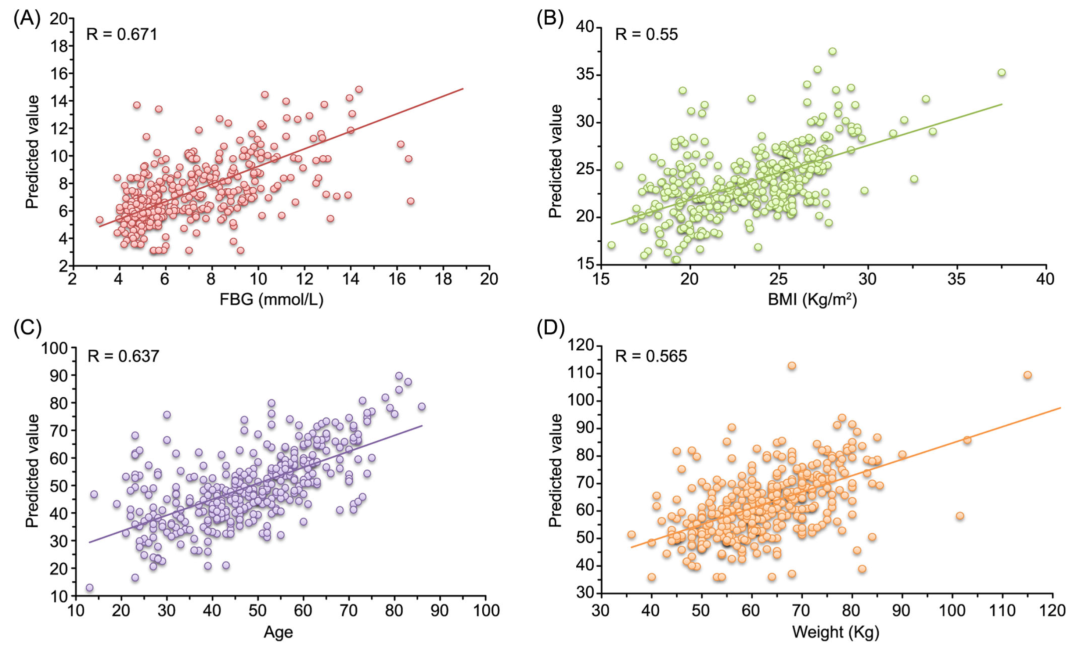
图 4. 在数据集D1上使用我们标记物种的神经网络的预测值和 T2D相关因素的实际值
(A) FBG、(B) BMI、(C) 年龄和 (D) 体重。每个 T2D相关因素的预测值的获得是通过基于 NN 的回归模型采用五折交叉验证法进行拟合。另外,我们通过统计计算获得真实值和预测值之间的 R 值(即 Pearson 线性相关系数)。BMI,体重指数;FBG,空腹血糖;T2D,2型糖尿病
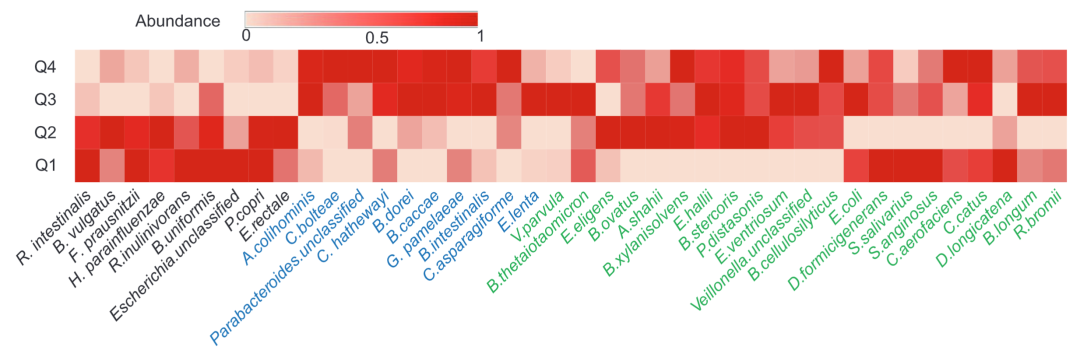
图 5. 空腹血糖四个动态区间中40个选定标记物种的平均相对丰度热图
动态区间由分位数统计确定(Q1:< 5.02;Q2:5.02–6.21;Q3:6.21–8.8;Q4:> 8.8)。在每个动态区间中,对相应样本的标记物种丰度进行平均。然后,对四个动态区间的每个标记物种的平均丰度进行归一化(映射到 [0, 1])。使用 Kruskal-Wallis 检验法分析标记物种的在所有样本间的变化情况,并依据此对的物种名称进行着色(黑色:健康样本中的丰度显着增加;蓝色:T2D 样本中的丰度显着增加;绿色:T2D 和健康样本之间的丰度没有显着差异)
讨 论
为了理解肠道微生物群在T2D发展中的作用并提供新的视角,我们在此提出了基于 NN 的框架来识别微生物标记物,这些标记物可用作肠道微生物群的代表,并在用于预测T2D中具有相对较高的预测性能。传统统计方法无法检测到的在病例和对照样本中没有显著变化的一些标记物,但相互作用网络分析和回归分析表明,这类标记物可能在人类肠道微生物组与T2D相关的改变中也起着至关重要的作用。该框架首先揭示了人类肠道微生物组中微生物标记物的 T2D 相关动态和相互作用,强烈表明标记物的累积效应可能是肠道微生物组改变的驱动因素。
深度学习方法通常需要大量样本进行训练和特征提取。尽管已经有了大量与 T2D相关的人类肠道微生物组研究,我们只考虑了全基因组测序生成的数据集作为本研究的条件。我们研究的主要限制是只有少量样本(N = 368,D1)来训练 NN 模型。尽管如此,通过确定合适的层数和节点数,我们的模型在5 fold CV 实验中对 T2D 相关样本进行分类方面获得了相对于其他方法更高的性能。由于数据集 D2 中缺乏对照样本(健康受试者),我们建立了数据集 D2+,包括随机选择的 D1 的 30% 对照样本作为对照,D2 的样本作为病例。当我们使用标记物种在数据集D2+ 上进行预测时,模型的预测性能 AUC 达到 76.2%,略低于在数据集D1上的预测结果。
分类结果表明,我们的NN模型的性能优于其他常规方法,包括SVM -Linear,SVM-RBF,RF和KNN。这表明我们的方法有较高可能性在复杂的微生物组数据集中揭示新型模式。然而,将NN模型应用于微生物组数据,由于其“黑匣子”特性仍然很具有挑战性,即通常很难解释哪些输入特征对输出结果起决定性作用。为了克服这一局限性,在本研究中,通过RF计算能区分病例和对照样品的特征(即物种或基因)的重要性,随后将NN模型作为分类器,依据不同特征子集的分类性能,确定哪个特征子集对于区分病例和对照样本是最重要的。我们的方法分别识别出了40种标记物种和90个标记基因。我们的NN模型使用这些生物标记物的预测性能稍好于使用所有微生物特征的预测性能。有趣的是,我们的大多数识别出的标记物种与先前的研究报道的与糖尿病有关标记物一致。例如,据报道,标记物种C. catus是短链脂肪酸的生产者(例如,丙酸,丁酸酯和丁酸酯),而Zhao等人的研究表明,短链脂肪酸产生的缺乏与T2D有关。在Jandhyala等人的研究观察到了从对照到慢性胰腺炎(CP)非糖尿病患者再到CP糖尿病患者的微生物组里R. bromii的丰度降低。Dewulf等研究人员发现C. aerofaciens的水平升高可能是与蛋白型果糖发酵有关的有益作用,该作用可用于控制包括糖尿病在内的相关代谢疾病。
我们的分析表明,所有在我们实验中的分类器使用我们方法识别出的标记物的预测性能比那些使用LEfSe识别出的生物标记物的预测性能更好。当使用 Kruskal-Wallis检验法分析病例和对照样本中所选生物标志物的丰度分布时,我们的识别出相当一部分标志物物种(n = 24)的丰度变化不大。因此,我们假设标记物种之间的相互作用,而不是单个物种的作用,会影响 T2D 相关的微生物组。为了解决这个问题,我们构建了有向生物标志物相互作用网络,这些网络提供了有关生物标志物如何相互影响的方向信息(图 2C、D 和 3C、D)。这些生物标志物相互作用证明了微生物标志物物种相互干扰的模式。例如,我们的标记物种相互作用网络表明E. coli 和S. salivarius几乎影响其他所有的标记物种,这表明它们可能是网络的主要驱动因素。同时,E. coli受到 B. thetaiotaomicron 和 S. salivarius 的影响;因此,B. thetaiotaomicron可以被认为是微生物群落的潜在间接驱动因素。这些标记物种之间的相互作用可能比单个物种携带更多的信息。在这方面,在病例和对照样本中相对丰度没有显着差异的标记物种可能在与 T2D 相关的肠道微生物改变中发挥特殊作用。Kruskal-Wallis 检验基于独立性假设,因此它不会考虑物种之间的相互作用,而我们的方法会考虑数据中的非平凡关系,因此会产生更好的答案。
回归分析表明,我们选定的生物标记物种所代表的人类肠道微生物组与FBG以及BMI,年龄和相应患者的体重这些因素有一定的相关性(P <0.05)。所有这些可能都是T2D的致病因素,但是就R和P值而言,FBG(r = 0.671,p = 4.87e -51;图4)与人类肠道微生物组的相关性最高。这一发现意味着,患者的肠道微生物组成可能会随着FBG的增加而不断改变T2D。我们根据其FBG值(即Q1:<5.02; Q2:5.02-6.21; Q3:6.21–8.8; Q4:> 8.8)将D1的样品分为四个区间,并绘制了各标记物种在每个区间内所有样品的平均丰度的热图。我们可以观察到,在所有样品中没有显着变化的24个标记物种,在FBG的四个区间中显示出不同的模式。我们的分析表明,包括E. coli 和 S. salivarius在内的这24个标记物种极大地影响了NN模型在对T2D样品分类中的性能,并且是相互作用网络中的关键物种。由于FBG在T2D的发展中逐渐增加,因此随着FBG的增加而改变的标记物种在驱动T2D相关的肠道微生物组改变中可能比使用基于统计量的方法确定的在病例和对照样品之间存在显著差异的物种更为重要。这些结果为理解糖尿病与人类肠道微生物组之间的关系提供了不同的观点,因此需要进一步研究。
结 论
本研究应用结合 NN 和 RF 的框架重新分析人类微生物组以识别 T2D 相关微生物标志物,而其中许多标志物被其他基于统计的方法所忽略。在验证和测试分析中,这些标志物被用来预测样本的疾病状态,从而可以用来解释人类肠道菌群在 T2D 发展过程中的微生物变化。标记的有向相互作用网络的构建捕获了与疾病相关的微生物群落的潜在驱动因素,这表明人类肠道微生物群的复杂性,其中许多核心物种相互作用而不是单个物种个体影响相关疾病。总之,使用 NN 模型和 RF,我们的分析产生了关于人类肠道微生物在T2D发展中的作用的新知识。
引文格式:
Shun guo, Haoran Zhang, Yunmeng Chun, Qingshan Jiang, Yingfei Ma. 2022. A neural network‐based framework to understand the type 2 diabetes‐related alteration of the human gut microbiome. iMeta. https://doi.org/10.1002/imt2.20
作者简介

郭顺(第一作者)
● 合成微生物组学研究中心助理研究员
● 2017年获得厦门大学博士学位。以第一作者发表多篇SCI论文(BMC bioinformatics, 2016, 2020; Peer J, 2022等)。研究方向为生物信息学,以及微生物学研究中的方法开发。

马迎飞(通讯作者)
● 研究员,博士生导师,合成微生物组学研究中心副主任
● 2009年获得中国科学院北京微生物研究所博士学位,先后在美国加州大学圣迭戈分校和纽约大学医学院从事人微生物组计划(Human Microbiome Project, HMP) 相关项目的研究主要研究。2015年入职深圳先进技术研究院。目前实验室承担国家各级研究项目包括:国自然面上、科技部重大研发专项、深圳市学科布局、技术攻关、孔雀团队等;近五年研究成果发表在Microbiome、Journal of Virology, Front. Microbiol, Scientific Reports 等国际权威期刊。课题组主要研究方向为:微生物组、噬菌体组、噬菌体合成生物学以及应用噬菌体防治耐药菌的应用研究
更多推荐
(▼ 点击跳转)
iMeta | 南昌大学丁霞等-水产养殖系统对中华鳖微生物组和肠道代谢组的影响
iMeta | 华中科大宁康组综述用于蛋白质结构预测的宏基因组定量分析
iMeta | 中科院李小方等膳食甘草促进小鼠镉解毒并调节肠道菌群代谢
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)

期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]
微信公众号
iMeta
责任编辑
微微