win10-R语言3.6.2-Bioconductor依赖管理&&KEGG富集分析&&通路图&&pathview报错解决
一、环境准备
下载安装包(64位),(如果没有翻墙,选择国内的源进行下载)链接如下:
https://mirrors.tuna.tsinghua.edu.cn/CRAN/bin/windows/base/R-3.6.2-win.exe
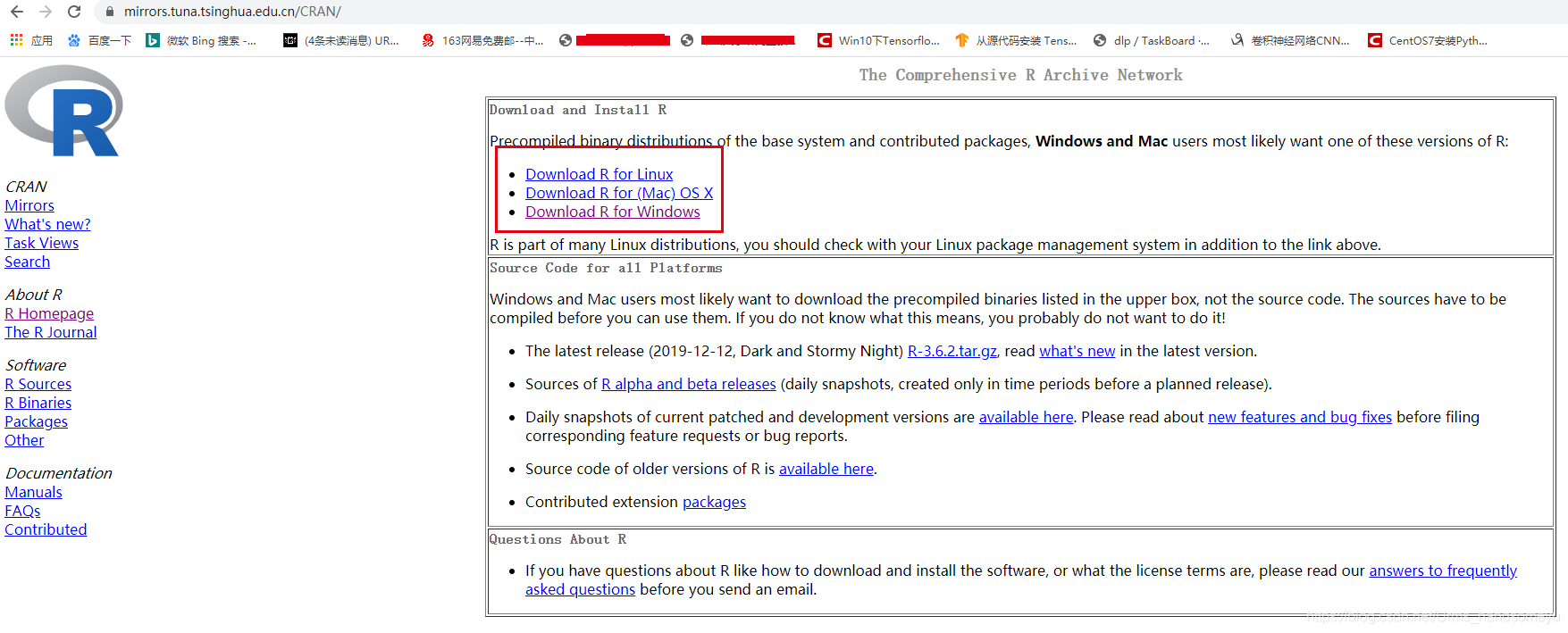
其他版本可以打开https://mirrors.tuna.tsinghua.edu.cn/CRAN,再根据自己物理机的配置自行选择,如下图所示:
或者选择本人的百度云链接进行下载,链接如下:
链接:https://pan.baidu.com/s/1_FEnaqRfSY1HpoQW9nfWbA
提取码:727u
安装
- 双击下载好的
R-3.6.2-win.exe文件,按提示安装即可。
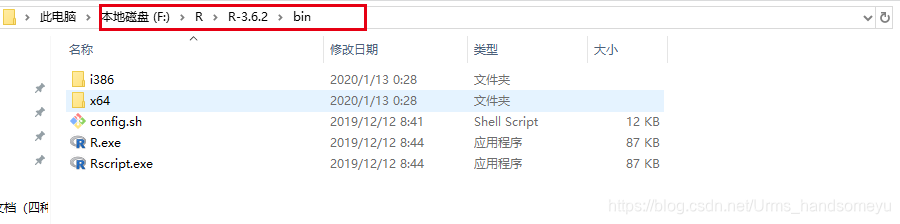
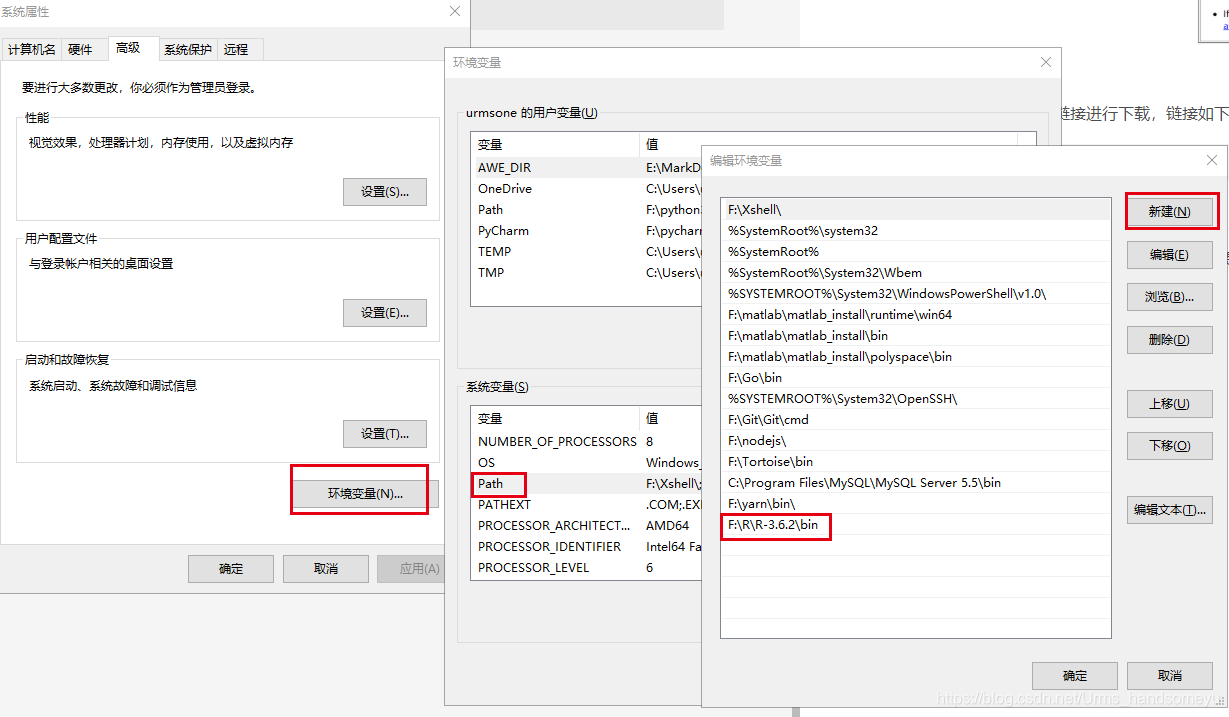
配置环境变量,在系统环境变量的Path中写入安装的R路径F:\R\R-3.6.2\bin
点击快捷方式即可运行程序
二、运行程序
代码:
#install.packages("colorspace")
#install.packages("stringi")
#options(BioC_mirror=”https://mirrors.tuna.tsinghua.edu.cn/bioc/“)
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install(version = "3.10")
#BiocManager::install(c("DOSE", "clusterProfiler","pathview"))
# setwd设置工作路径,文件的读入和写出都是相对与该路径
setwd("C:\\Users\\urmsone\\Desktop\\Li")
library("clusterProfiler")
rt=read.table("id.txt",sep="\t",header=T,check.names=F)
rt=rt[is.na(rt[,"entrezID"])==F,]
geneFC=rt$logFC
gene=rt$entrezID
names(geneFC)=gene
#kegg富集分析
kk <- enrichKEGG(gene = gene, organism = "hsa", pvalueCutoff =0.05, qvalueCutoff =0.05)
write.table(kk,file="KEGG.txt",sep="\t",quote=F,row.names = F)
#柱状图
tiff(file="barplot.tiff",width = 30,height = 20,units ="cm",compression="lzw",bg="white",res=600)
barplot(kk, drop = TRUE, showCategory = 20)
dev.off()
#点图
tiff(file="dotplot.tiff",width = 30,height = 20,units ="cm",compression="lzw",bg="white",res=600)
dotplot(kk, showCategory = 20)
dev.off()
#通路图
library("pathview")
source("./pathview-u.R")
keggxls=read.table("KEGG.txt",sep="\t",header=T)
for(i in keggxls$ID){
pv.out <- pathview(gene.data = geneFC, pathway.id = i, species = "hsa", out.suffix = "pathview")
}
PS:首次运行时才需要安装依赖
安装依赖:
# 该程序需要手动安装的依赖有colorspace,stringi,DOSE,clusterProfiler,pathview
install.packages("colorspace")
install.packages("stringi")
# 换源
options(BioC_mirror=”https://mirrors.tuna.tsinghua.edu.cn/bioc/“)
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.10")
BiocManager::install(c("DOSE", "clusterProfiler","pathview"))
三、运行程序
打开R客户端

将代码复制进去执行
setwd("C:\\Users\\urmsone\\Desktop\\Li")
library("clusterProfiler")
rt=read.table("id.txt",sep="\t",header=T,check.names=F)
rt=rt[is.na(rt[,"entrezID"])==F,]
geneFC=rt$logFC
gene=rt$entrezID
names(geneFC)=gene
#kegg富集分析
kk <- enrichKEGG(gene = gene, organism = "hsa", pvalueCutoff =0.05, qvalueCutoff =0.05)
write.table(kk,file="KEGG.txt",sep="\t",quote=F,row.names = F)
#通路图
library("pathview")
#source("./pathview-u.R")
keggxls=read.table("KEGG.txt",sep="\t",header=T)
for(i in keggxls$ID){
pv.out <- pathview(gene.data = geneFC, pathway.id = i, species = "hsa", out.suffix = "pathview")
}
PS: 如果输出的KEGG.txt文件中含有ID为hsa04251的数据,可能会报错。报错在下节讲解如何解决
四、报错解决
报错: With R version 3.5 or greater, install Bioconductor packages using BiocManager
-
执行
source("http://bioconductor.org/biocLite.R")报错错误: With R version 3.5 or greater, install Bioconductor packages using BiocManager; see https://bioconductor.org/install
-
原因:R 3.6以后使用BoicManager进行依赖管理
-
解决
# R 3.6以后使用BoicManager进行依赖管理 options(BioC_mirror=”https://mirrors.tuna.tsinghua.edu.cn/bioc/“) if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(version = "3.10") # 要安装什么依赖直接BiocManager::install("xxx")即可
报错:只有负下标里才能有零
-
执行pv.out <- pathview(gene.data = geneFC, pathway.id = i, species = “hsa”, out.suffix = “pathview”)报错
Info: Writing image file hsa04215.pathview.png Info: some node width is different from others, and hence adjusted! Error in img[pidx[i, 3]:pidx[i, 4], sel.px, 1:3] : 只有负下标里才能有零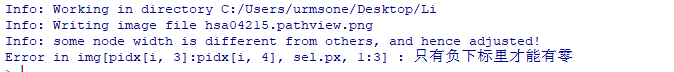
-
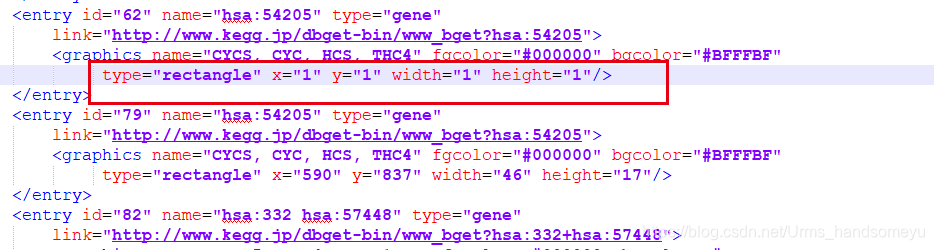
原因:拉下来的hsa04215.xml文件中部分entry为
type="rectangle" x="1" y="1" width="1" height="1",node.info函数在解析该xml文件的时候会生成x=1 y=1的记录,导致生成png图片的时候报错
-
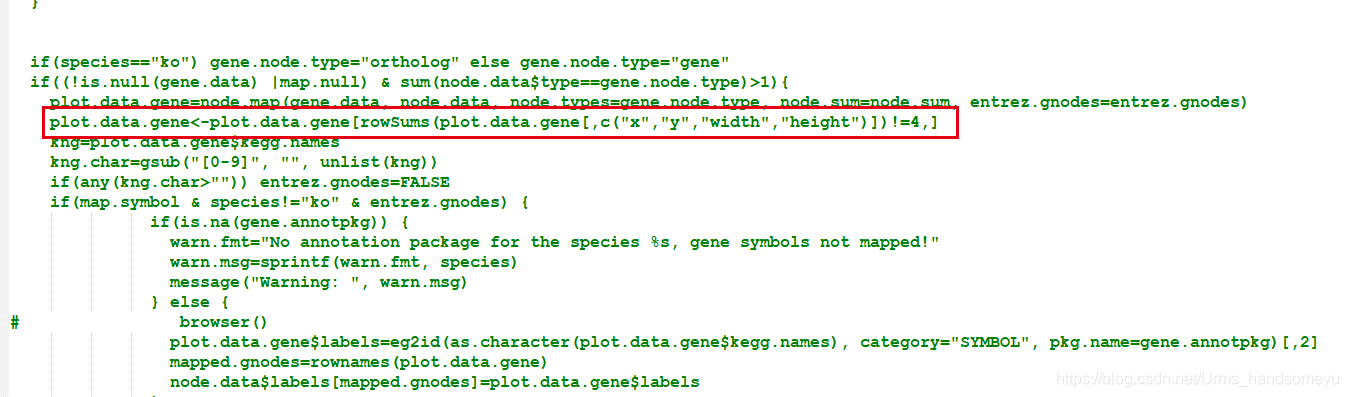
解决: 在pathview函数中把entry为
type="rectangle" x="1" y="1" width="1" height="1"的数据过滤掉
- 首先,导入pathview
> library("pathview") 载入需要的程辑包:org.Hs.eg.db 载入需要的程辑包:AnnotationDbi 载入需要的程辑包:stats4 载入需要的程辑包:BiocGenerics 载入需要的程辑包:parallel ... - 然后获取pathview的源码
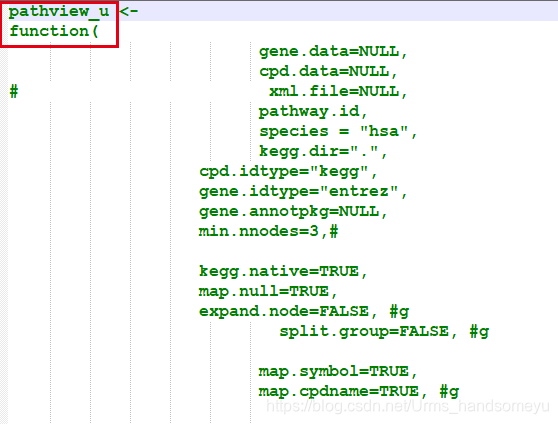
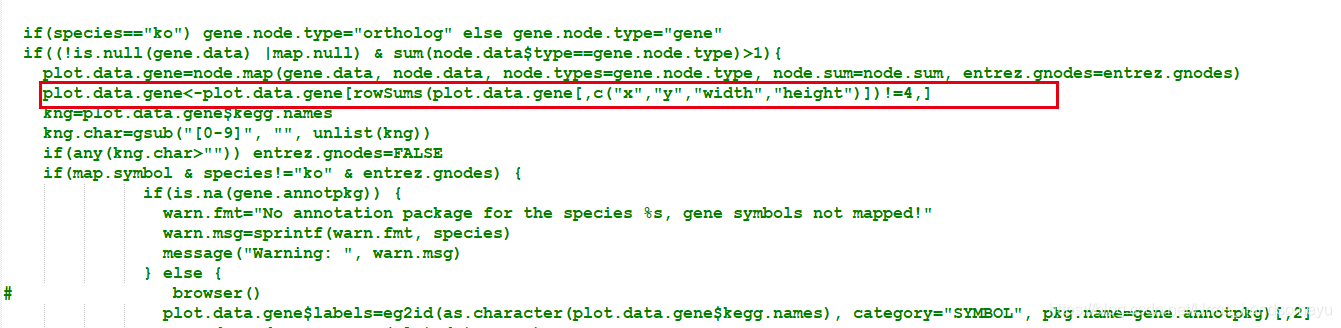
> pathview function (gene.data = NULL, cpd.data = NULL, pathway.id, species = "hsa", kegg.dir = ".", cpd.idtype = "kegg", gene.idtype = "entrez", gene.annotpkg = NULL, min.nnodes = 3, kegg.native = TRUE, map.null = TRUE, expand.node = FALSE, split.group = FALSE, map.symbol = TRUE, map.cpdname = TRUE, node.sum = "sum", discrete = list(gene = FALSE, cpd = FALSE), limit = list(gene = 1, cpd = 1), bins = list(gene = 10, cpd = 10), both.dirs = list(gene = T, cpd = T), trans.fun = list(gene = NULL, cpd = NULL), low = list(gene = "green", cpd = "blue"), mid = list(gene = "gray", cpd = "gray"), high = list(gene = "red", cpd = "yellow"), na.col = "transparent", ...) - 新建一个pathview-u.R文件,将pathview函数的源码写入,并修改函数名为pathview-u,在
plot.data.gene=node.map..后加入plot.data.gene<-plot.data.gene[rowSums(plot.data.gene[,c("x","y","width","height")])!=4,]
- 首先,导入pathview
-
导入修改后的函数,并使用:
> source("./pathview-u.R") > for(i in keggxls$ID){ + pv.out <- pathview_u(gene.data = geneFC, pathway.id = i, species = "hsa", out.suffix = "pathview") + } Info: Downloading xml files for hsa04657, 1/1 pathways.. Info: Downloading png files for hsa04657, 1/1 pathways.. 'select()' returned 1:1 mapping between keys and columns Info: Working in directory C:/Users/urmsone/Desktop/Li Info: Writing image file hsa04657.pathview.png Info: Downloading xml files for hsa04668, 1/1 pathways.. Info: Downloading png files for hsa04668, 1/1 pathways.. 'select()' returned 1:1 mapping between keys and columns Info: Working in directory C:/Users/urmsone/Desktop/Li Info: Writing image file hsa04668.pathview.png Info: Downloading xml files for hsa04066, 1/1 pathways.. Info: Downloading png files for hsa04066, 1/1 pathways.. 'select()' returned 1:1 mapping between keys and columns Info: Working in directory C:/Users/urmsone/Desktop/Li Info: Writing image file hsa04066.pathview.png ...
报错:Error in readPNG(paste(kegg.dir, "/", pathway.name, ".png", sep = "")) : libpng error: Read Error
五、Demo获取链接
完整Demo和使用说明请参考https://github.com/UrmsOne/KEGG-demo
