
写在前面
后台难得有读者私信,请教了下图中文章的GSEA图能不能用R来画,今天就来简单写个教学。


GSEA(Gene Set EnrichmentAnalysis),即基因集富集分析,它的基本思想是使用预定义的基因,将基因按照在两类样本中的差异表达程度排序,然后检验预先设定的基因集合是否在这个排序表的顶端或者底端富集。
GSEA 和GO、KEGG pathway不同的地方在于,后两者会提前设定一个阈值,只关注差异变化大的基因(相当于重点班)。这样子容易遗漏部分差异表达不显著却有重要生物学意义的基因(成绩一般,但是很有天赋)。所以GSEA分析比较适用于,传统分析方法筛选后样本过少的数据集。
数据准备
直接用之前的转录组差异分析后的数据来演示,数据格式如下:

至于基因差异分析怎么l做,有好多种办法,常用的有DESeq2、EdgeR、limma,有问题的话可以私信~因为GSEA只需要SYMBOL(基因名)和foldchange (或logFC)两列,所以可以把不需要的删掉。(以上操作在EXCEL,或者用R的tidyverse(数据框处理可以参考文章: )进行操作都可以,怎么方便怎么来)
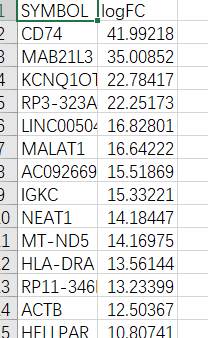
开始分析
安装并导入要用到的R包
BiocManager::install("clusterProfiler") #感谢Y叔的clusterprofiler包
BiocManager::install("enrichplot") #画图需要
BiocManager::install("org.Hs.eg.db") #基因注释需要
library(org.Hs.eg.db)
library(clusterProfiler)
library(enrichplot)
导入数据
setwd("D:/Note/MZBJ/Q_A") #设置文件所在位置
df = read.table("gene_diff.txt",header = T) #读入txt
# df = read.csv("gene_diff.csv",header = T) #读入csv
head(df)#查看前面几行
dim(df)#数据总共几行几列
> head(df)#查看前面几行
SYMBOL logFC
1 CD74 41.99218
2 MAB21L3 35.00852
3 KCNQ1OT1 22.78417
4 RP3-323A16.1 22.25173
5 LINC00504 16.82801
6 MALAT1 16.64222
> dim(df)#数据总共几行几列
[1] 5057 2
转换基因ID
如基因名是symbol,需要将基因ID转换为Entrez ID格式。Entrez ID实际上是指的Entrez gene ID,是对应于染色体上一个gene location的。每一个发现的基因都会被编制一个统一的编号,而Entrez ID是指的来自于NCBI旗下的Entrez gene数据库所使用的编号。因为Entrez ID具有特异性,所以后续分析更适合用Entrez ID。
df_id<-bitr(df$SYMBOL, #转换的列是df数据框中的SYMBOL列
fromType = "SYMBOL",#需要转换ID类型
toType = "ENTREZID",#转换成的ID类型
OrgDb = "org.Hs.eg.db")#对应的物种,小鼠的是org.Mm.eg.db
>'select()' returned 1:many mapping between keys and columns
Warning message:
In bitr(df$SYMBOL, fromType = "SYMBOL", toType = "ENTREZID", OrgDb = "org.Hs.eg.db") :
7.87% of input gene IDs are fail to map... #7.87%没有比对到就是没有转换成功
把两个数据框df 和 df_id根据SYMBOL列合并。
df_all<-merge(df,df_id,by="SYMBOL",all=F)#使用merge合并
head(df_all) #再看看数据
dim(df_all) #因为有一部分没转换成功,所以数量就少了。
> head(df_all)
SYMBOL logFC ENTREZID
1 A2M -0.713519723 2
2 AAK1 -0.089497971 22848
3 AAMP -0.014536797 14
4 AARS2 0.077105219 57505
5 AASDHPPT -0.000560858 60496
6 ABCA1 0.436678052 19
> dim(df_all)
[1] 4660 3
GAEA
df_all_sort <- df_all[order(df_all$logFC, decreasing = T),]#先按照logFC降序排序
gene_fc = df_all_sort$logFC #把foldchange按照从大到小提取出来
head(gene_fc)
names(gene_fc) <- df_all_sort$ENTREZID #给上面提取的foldchange加上对应上ENTREZID
head(gene_fc)
> head(gene_fc)
[1] 41.99218 35.00852 22.78417 16.82801 16.64222 15.33221
> head(gene_fc)
972 126868 10984 201853 378938 3514
41.99218 35.00852 22.78417 16.82801 16.64222 15.33221
准备以上的东西,接下来一行代码解决。
#以KEGG Pathway示例
KEGG <- gseKEGG(gene_fc, organism = "hsa") #具体参数在下面
> KEGG <- gseKEGG(gene_fc, organism = "hsa")
Reading KEGG annotation online:
Reading KEGG annotation online:
preparing geneSet collections...
GSEA analysis...
leading edge analysis...
done...
Warning messages:
1: In preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, :
There are ties in the preranked stats (0.13% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
2: In serialize(data, node$con) : 载入时'package:stats'可能无用
3: In serialize(data, node$con) : 载入时'package:stats'可能无用
4: In serialize(data, node$con) : 载入时'package:stats'可能无用
5: In serialize(data, node$con) : 载入时'package:stats'可能无用
6: In serialize(data, node$con) : 载入时'package:stats'可能无用
7: In serialize(data, node$con) : 载入时'package:stats'可能无用
8: In fgseaMultilevel(...) :
For some pathways, in reality P-values are less than 1e-10. You can set the `eps` argument to zero for better estimation.
如果要做GO富集呢?
#GO富集
GO <- gseGO(
gene_fc, #gene_fc
ont = "BP",# "BP"、"MF"和"CC"或"ALL"
OrgDb = org.Hs.eg.db,#人类注释基因
keyType = "ENTREZID",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",#p值校正方法
)
#KEGG富集
gseKEGG(
geneList,
organism = "hsa",
keyType = "kegg",
exponent = 1,
minGSSize = 10,
maxGSSize = 500,
eps = 1e-10,
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
verbose = TRUE,
use_internal_data = FALSE,
seed = FALSE,
by = "fgsea",
...
)
head(KEGG)#看一下这个文件
> head(KEGG)
ID Description setSize enrichmentScore NES
hsa03010 hsa03010 Ribosome 99 -0.8707285 -2.370839
hsa05152 hsa05152 Tuberculosis 87 0.8678558 1.786981
hsa05171 hsa05171 Coronavirus disease - COVID-19 142 -0.5976011 -1.704522
hsa04512 hsa04512 ECM-receptor interaction 19 -0.8866402 -1.913989
pvalue p.adjust qvalues rank leading_edge
hsa03010 0.0000000001 0.0000000257 2.431579e-08 289 tags=65%, list=6%, signal=62%
hsa05152 0.0002124294 0.0272971804 2.582695e-02 279 tags=30%, list=6%, signal=29%
hsa05171 0.0004376904 0.0290749106 2.750893e-02 289 tags=46%, list=6%, signal=45%
hsa04512 0.0004525278 0.0290749106 2.750893e-02 250 tags=58%, list=5%, signal=55%
core_enrichment
hsa03010 6231/6193/4736/6235/2197/6218/6166/6167/6157/3921/6129/140801/6152/6125/6169/6124/9349/6141/6138/6187/6228/6144/6135/6202/6155/6154/6132/6160/6159/6147/6156/6210/6230/6175/6122/6128/11224/23521/9045/25873/6161/6201/6208/6189/6181/6188/6133/6165/6194/6139/6168/6224/6143/6142/6222/6164/6176/6232/6206/6223/6171/6233/6134/6137
hsa05152
简单解释一下结果的意思:
ID代表KEGG中的信号通路Description对信号通路的描述setSize该信号通路的基因个数enrichmentScore富集分数,也就是ESNES标准化以后的ES,全称normalized enrichment score、qvalues,或者说FDR q-val(false discovery rate)错误发现率rank排名core_enrichment,富集该目的通路的基因列表。
sortKEGG<-KEGG[order(KEGG$enrichmentScore, decreasing = T),]#按照enrichment score从高到低排序
head(sortKEGG)
dim(sortKEGG)
write.table(sortKEGG,"gsea_sortKEGG.txt") #保存结果
结果可视化
#gseaplot2用法
gseaplot2(
x, #gseaResult object,即GSEA结果
geneSetID,#富集的ID编号
title = "", #标题
color = "green",#GSEA线条颜色
base_size = 11,#基础字体大小
rel_heights = c(1.5, 0.5, 1),#副图的相对高度
subplots = 1:3, #要显示哪些副图 如subplots=c(1,3) #只要第一和第三个图,subplots=1#只要第一个图
pvalue_table = FALSE, #是否添加 pvalue table
ES_geom = "line" #running enrichment score用先还是用点ES_geom = "dot"
)
先来个常规的
gseaplot2(KEGG, "hsa05152", color = "firebrick", rel_heights=c(1, .2, .6))

再来个文章里的:
paths <- c("hsa03010", "hsa05152", "hsa05171", "hsa04512")#选取你需要展示的通路ID
gseaplot2(KEGG,paths, pvalue_table = TRUE)

gseaplot2(KEGG,paths,color = colorspace::rainbow_hcl(4),subplots=c(1,2), pvalue_table = TRUE)
#换个颜色,只显示上面两个副图

剩下的一点细节用AI处理一下就可以~
以上教程为本人的粗浅记录,如有错误还请各位读者指正如果觉得有用,希望大家多多点赞分享
代码领取:公众号后台私信“GSEA"即可领取全部代码。
往期内容: