一.背景
三阴性乳腺癌(TNBC)是一种侵袭性亚型,其特点是广泛的瘤内异质性。三阴性乳腺癌在临床上被定义为缺乏雌激素受体(ER)和孕激素受体(PgR)表达以及人类表皮生长因子受体2(HER2)基因扩增,占所有乳腺癌的20%,与其他乳腺癌亚型相比,其临床过程更具侵略性。少数研究在单细胞水平上描述了TNBC的基因组多样性,揭示了一种模式,反映了TNBC进展过程中拷贝数变异的点状进化,然后是优势亚克隆的扩张。虽然这些发现意味着这些亚克隆具有驱动其选择优势的特性,但仅仅基于DNA的分析还不能阐明支撑这一过程的细胞状态和命运。
二.方法
我们对6个新收集的、未经治疗的原发性TNBC肿瘤中的1500个细胞进行了单细胞RNA测序。
三 .结果
1 获取原发性TNBC的scRNA-seq图谱
为了了解TNBC的细胞间异质性,我们收集了6名妇女的肿瘤,这些妇女在接受任何局部或全身治疗之前,出现了原发性、非转移性的三阴性浸润性导管癌。新鲜的肿瘤进行了快速分离,然后用流式细胞仪对有活力的单细胞进行分类。为了捕捉到肿瘤细胞组成的全部光谱,我们在没有预选的情况下对细胞和肿瘤的一个子集进行了分选,为了确保有足够数量的恶性细胞用于分析,我们在基于CD45染色的免疫细胞耗尽后对另一个子集进行了分选(图1a)。各个细胞进行了cDNA的制备和文库的构建,然后进行下一代测序(NGS)。经过严格的质量控制和归一化,我们共分析了1189个细胞,患者中从78个细胞(肿瘤58)到286个细胞(肿瘤89)不等(图1a)。
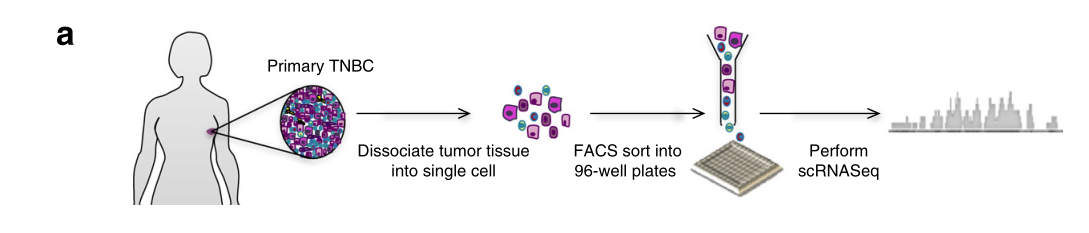
工作流程显示收集和处理新鲜TNBC原发肿瘤以生成scRNA-seq数据
2 原发性TNBC内的细胞异质性
我们的第一项分析是利用涉及标记基因和聚类的多步骤方法确定肿瘤内的不同细胞群(图1b,c)。这种组合方法旨在提供一个比单独使用任何一种方法都更强大的细胞类型识别。因此,我们首先评估了以前建立的特定基因组的表达,以定义免疫、内皮和基质细胞,以及关键的乳腺上皮亚群,包括基底细胞、腔内祖细胞和成熟腔内细胞。然后,我们用聚类法来完善所确定的细胞类型。这种综合分析将1189个细胞中的1112个可靠地分为非上皮细胞(n = 244)和上皮细胞(n = 868)类型。
众所周知,TNBC的一个子集表现出大量的免疫细胞浸润,正如预期的那样,未经CD45选择的肿瘤含有很大比例的免疫细胞,其中大部分是T淋巴细胞。下一个最普遍的免疫细胞亚群是巨噬细胞,在每个CD45未选择的肿瘤中都存在。与免疫细胞一样,已知基质元素在不同的TNBC病例中有所不同,我们确定这种细胞在每个肿瘤中占总细胞的15%以下。
内皮细胞是一个少数群体,在一个肿瘤中最多占4%的细胞(图1c)。每个肿瘤中最突出的上皮细胞群表达了腔内细胞和腔内祖细胞的标记,而在一些肿瘤中(如89号肿瘤),少数细胞群明显表达了肌上皮细胞的标记,包括ACTA2和TAGLN(图1b)。
作为识别恶性细胞的第一步,我们使用先前用于识别G1/S和G2/M细胞周期阶段并区分高周期和低周期细胞的验证基因标记来确定它们的细胞周期状态。该分析显示,肿瘤中循环细胞的比例存在很大差异,范围从<5%(58例肿瘤)到>34%(81例肿瘤,图1d)。值得注意的是,大多数循环细胞(占所有循环细胞的98.5%)被鉴定为上皮细胞,这与我们的细胞类型分类一致,并表明恶性细胞位于此隔室(图1e)。我们用Ki67(一种广泛使用的循环细胞临床标记物)对肿瘤切片进行免疫组化染色,验证了这些发现。事实上,我们观察到预测的循环细胞比例与每个肿瘤中ki67阳性细胞的百分比之间存在高度相关性(图1e)。

b通过质量控制的1189个细胞的热图,列表示细胞,行表示左侧所示细胞类型的已建立的基因表达标记,分别为6例TNBC病例的每一例聚类。上面的柱状图表示推断出的高循环(粉红色)和低循环(灰色)细胞,通过量化一组相关基因的表达来识别。底部条形图表示存在/不存在CD45 +细胞耗竭情况下收集的细胞。
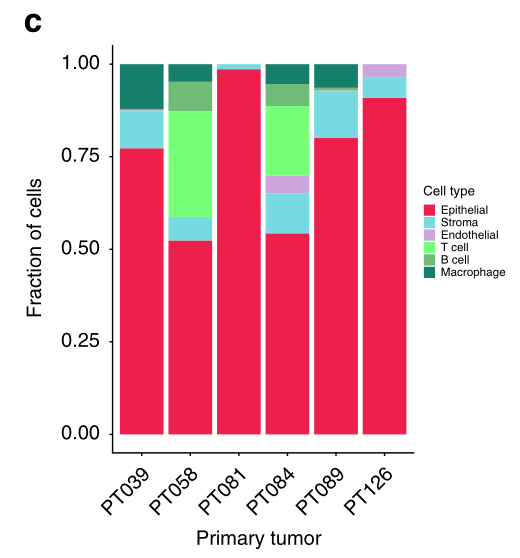
c 柱状图描述了按病人分配到特定细胞类型的1112个细胞的分布。
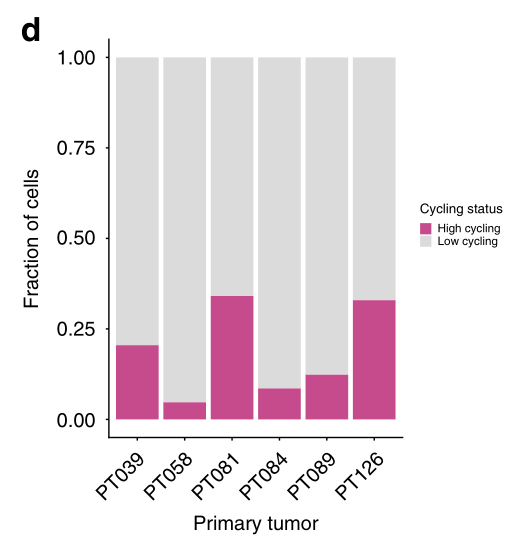
d 条形图描述了1112个细胞的高循环/低循环分布,按病人分类。
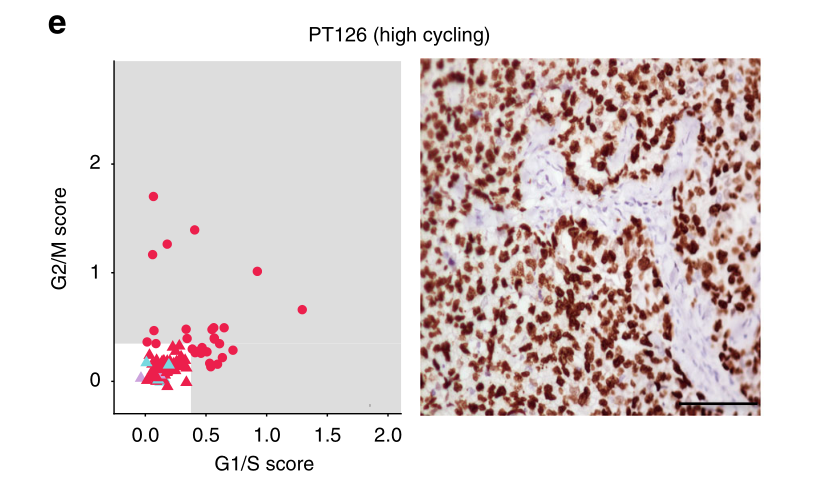
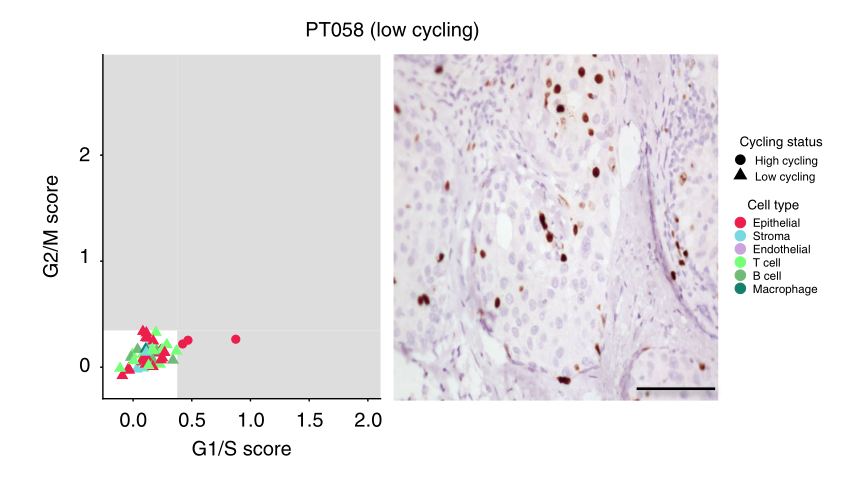
e 两个有代表性的TNBC患者的增殖特征,描述为单细胞的推断循环状态(左)或Ki67的免疫组化染色(右)。比例尺代表50微米。如果一个细胞具有较高的G1/S或G2/M分数,则被认为是高周期性的,这是通过量化一组相关基因的表达而确定的。这两种量化增殖的方法显示出良好的一致性
3亚克隆异质性定义了恶性的TNBC细胞
为了进一步支持恶性细胞与非恶性细胞的分类,我们使用了基于(i)基因表达聚类、(ii)估计拷贝数变异(CNVs)和(iii)转录组细胞间异质性的互补方法。使用tSNE聚类分析患者的所有细胞,发现非上皮细胞群和上皮细胞群之间有很大的区别,前者根据其细胞类型被很好地分隔成不同的集群,后者则形成多个亚群(图2a)。当非上皮细胞和上皮细胞被分别分析时,这种模式最为明显(图2b,c)。
与非上皮细胞相比,上皮细胞一般分为肿瘤特定的集群(特别是39号和81号肿瘤),但有趣的是,也有来自多个肿瘤的细胞组成的集群(图2a-c)。之前对黑色素瘤和胶质母细胞瘤的单细胞分析显示,恶性细胞的集群主要是由病人组成的,这支持了肿瘤间异质性的显著程度。相反,与我们的研究结果相一致,最近对乳腺癌的单细胞分析显示,恶性细胞既有病人特异性的,也有共同的集群,暗示了乳腺癌特殊的瘤内异质性和存在由共同状态定义的亚群。
然后,我们试图通过使用先前描述的方法识别从单细胞基因表达谱推断的大规模 CNV,来进一步定义上皮细胞群中的恶性细胞。出于标准化目的,我们使用了来自正常乳腺上皮细胞的单细胞基因表达数据。先前的单细胞基因组研究证实,TNBC 表现出高度可变的 CNV 模式,部分是由间断的肿瘤基因组进化驱动的。因此,尽管一些 TNBC 表现出以大 CNV 为特征的亚克隆,但许多肿瘤中的亚克隆表现出相对较小的 CNV,超出了转录组数据推断的分辨率 。正如预测的那样,我们发现亚克隆大规模 CNV 在一些(特别是肿瘤 39 和 81)中很明显,但不是所有肿瘤,并且没有肿瘤显示出所有细胞共有的 CNV 克隆模式(图 2d)。然后,我们通过批量全外显子组测序(WES,图 2e)验证了这些发现,这证明了单细胞转录组数据推断的 CNV 与基因组数据中证明的 CNV 之间的高度一致性(图 2d,e) ).例如,对于 39 号和 81 号肿瘤,这两种方法都明显增加了 1 号染色体; 5 号染色体的明显丢失和 12 号染色体的增加在 39 号肿瘤中得到证实,而 8 号染色体的增加在 81 号肿瘤中很明显。此外,89 号肿瘤表明没有推断的克隆或亚克隆获得或丢失,也显示出很少的拷贝WES 号码变更。我们还发现,诊断时原发肿瘤的大小与显性亚克隆的存在相关,这在 scRNAseq 推断的 CNV 和大量 WES 中都很明显。例如,肿瘤 39(9.5 cm 肿瘤)表现出显性亚克隆群体,而肿瘤 89(1.5 cm 肿瘤)几乎没有这种群体的证据(图 2d,e)。尽管肿瘤数量很少,但这一发现扩展了我们之前记录稳定的非整倍体重排的单细胞基因组分析,并表明这种改变在乳腺癌的克隆扩增过程中控制转录 。
由于某些肿瘤中不存在克隆性 CNV,因此无法将每个上皮细胞归类为良性或恶性,接下来我们研究了肿瘤上皮细胞群内部和之间的转录异质性与正常原发性乳腺上皮细胞之间的异质性 19(图 2f)。根据最近对其他肿瘤类型的单细胞分析结果,我们推断非恶性上皮细胞将高度一致,亚克隆 CNV 定义的肿瘤上皮细胞群也是如此,而其余恶性细胞可能是异质的,因此不一致 13 .正如预期的那样,正常上皮细胞显示出相当高的一致性(平均 Spearman 相关性 0.38),亚克隆 CNV 定义的患者 39 和 81 的细胞亚组也是如此(肿瘤 39 的 Spearman 相关性为 0.45,肿瘤 81 的相关性为 0.41),这也被确定为不同的上皮细胞簇(图 2c)。相比之下,剩余的肿瘤上皮细胞通常在肿瘤内部和肿瘤之间表现出较弱的一致性,支持它们的异质性,因此它们可能是恶性细胞(Spearman 相关性:患者 89 为 0.25,患者 84 为 0.26,患者 58 和 126 为 0.30)(图.2f).总之,这些数据表明这些肿瘤内的上皮细胞的特征是反映亚克隆 CNV 的表达模式,或缺乏 CNV 和相应的高度细胞间异质性。这些发现和几乎完全存在于上皮隔室内的循环细胞共同支持了这样的假设,即大多数已鉴定的上皮细胞是恶性细胞。
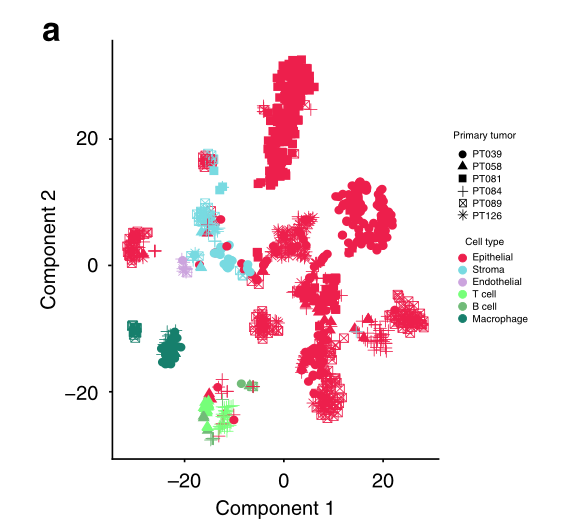
a 所有 1112 个分类细胞的 t-SNE 图,显示按细胞类型分离非上皮细胞。

b 244个非上皮性细胞的t-SNE图,显示按细胞类型分离,没有明显的患者效应。
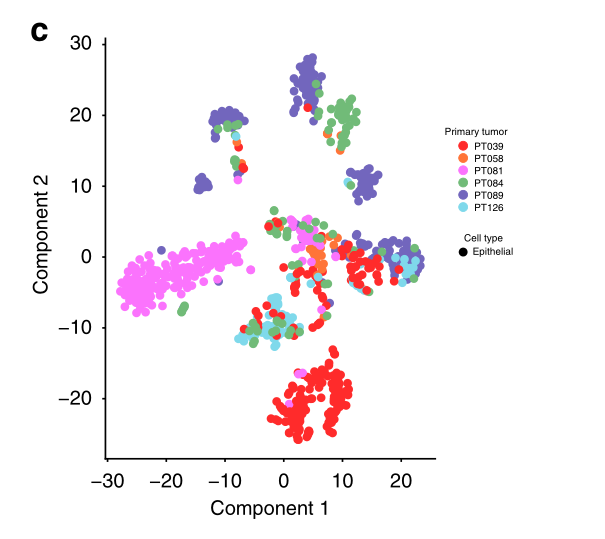
c 868上皮细胞的t-SNE图,显示患者的混合分离,以及来自不同患者的细胞的大量聚集,表明肿瘤内存在明显的异质性。

d 从单细胞基因表达数据推断CNV。列代表单个细胞,行代表一组选定的基因,按照它们的基因组坐标排列(染色体编号显示在左边)。左侧显示一组240个正常乳腺上皮细胞以供比较,所有TNBC病例的上皮细胞分别为每个患者聚集在一起。扩增(红色)或缺失(蓝色)是通过计算每个基因的100个基因移动平均表达分数来推断的,以感兴趣的基因为中心。肿瘤39和81中由共享CNV定义的显著亚克隆由顶部的括号表示(“克隆性”).
e 6例TNBC病例中4例的E-WES数据与从单细胞转录本推断的CNV呼唤高度一致(d)。基因组坐标从上到下按d排列,每个区域的平均拷贝数(“CNV均值”)在连续的标度上表示,红色表示获得,蓝色表示丢失。因此,从左(d)到右(e)的扫描允许对相同区域的推断CNV(d)和实际CNV(E)进行比较。
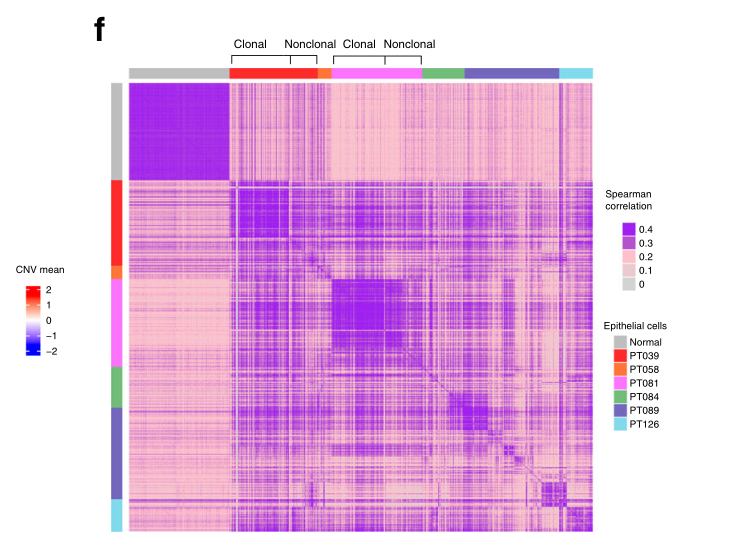
f 正常上皮细胞和TNBC上皮细胞的表达谱之间的相关性图从左到右与d.正常细胞以及肿瘤39和81的共享CNV定义的恶性克隆性亚群(顶部表示为克隆)之间的相关图是相关的。所有肿瘤中剩余的非克隆性上皮细胞群体的相关性相对较差,支持它们是恶性细胞。
4 共有的恶性亚群反映了不同的表型
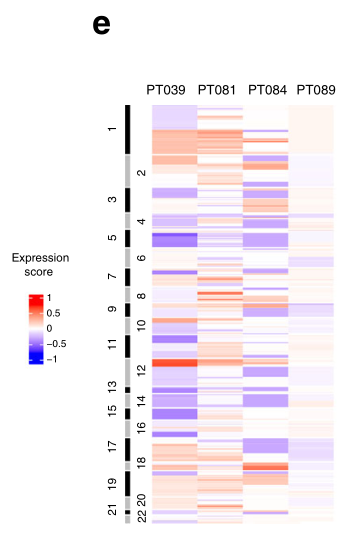
由于我们的聚类分析表明,上皮细胞亚群共享共同的转录特征,但来自多个肿瘤(图2c),我们接下来试图通过对所有上皮细胞的聚集来揭示这些群体的共同生物学,同时通过线性回归排除患者特有的影响。使用这种方法,我们识别了五个细胞簇,其中一个(簇2)由所有肿瘤中相当大比例的细胞代表,另一个(簇3)存在于六个肿瘤中的五个(图3a,b)。簇4在缺少簇3细胞的肿瘤81中最为突出,而簇1和簇5总共代表<70个细胞,仅在肿瘤84和89中存在(图3b)。重要的是,群2中的大多数细胞(55%)含有大规模的CNV,因此确定该群由恶性细胞组成。因此,组2包含最高比例的高周期细胞(40%),而组3和4也包含显著的高周期群体(分别为11%和13%),而组1和5不包含(1个高周期细胞/组)。结合图2中的分析,这些发现支持第2、3和4组的恶性身份,而第1组和第5组中的少量细胞由于其较少的增殖表型而不能被确定为恶性细胞。
然后,我们研究了恶性细胞群和单个肿瘤,以丰富来自已建立的基础、腔前体细胞(LP)和成熟腔(ML)细胞的批量RNA-SEQ的基因表达特征(图3b,c)。构成大多数恶性细胞的第2类和第4类与LP特征的关联度最高,这与支持LP作为乳腺癌起源细胞的数据保持一致。相比之下,第3群通过ML细胞签名明显区分开来。因此,我们发现,带有恶性表型的不同的周期细胞亚群的特征是表达谱反映了沿腔上皮谱系的分化。
接下来,我们使用一组来自对乳腺肿瘤的非监督分析的基因表达特征来研究聚类亚群。这些肿瘤特异性特征包括TNBCtype-4特征,它定义了四个TNBC亚型的某些临床和生物学特征,以及固有基本特征,它是通过比较乳腺癌亚型(ER+、HER2+和TNBC)而建立的。本征基底肿瘤代表大多数TNBCs;它们与多种TNBC型亚型重叠,与非本征基底TNBCs相比克隆异质性增加(图3d)。我们发现,簇2亚群与单一的TNBC型特征相关性最强,称为“Basal Like 1”,它富含细胞周期和DNA修复基因,与该簇中存在的高比例周期细胞一致(图3e)。该簇也是本征基本特征信号最丰富的(图3d)。簇4也富含Basal-like 1特征,而簇3的TNBC型“腔雄激素受体”特征富集度最高,与这种分化的腔(ML)特征簇的丰富一致(图3b-e)。因此,簇群亚群在其已建立的TNBC表型的表达上是不同的。值得注意的是,在图3a中,团簇2和3的两个潜在的“亚团簇”是显而易见的。对这些亚群应用多个标记表明,它们并不总是与各自的主群不同,尽管群3的亚群确实表现出比群3作为一个整体更成熟的流明特征。
对每个肿瘤进行这种分析,我们发现81号肿瘤主要是由对应于LP-like 2和4群的Basal-like 1细胞组成,并且循环细胞的比例最高(31%)。这一发现与CNV和细胞间异质性分析所记录的该肿瘤的相对同质性一致(图2d-f)。相比之下,84和89号肿瘤包括基底样1/LP细胞,但也有大量分化程度更高的管腔样细胞亚群,表达管腔雄性激素受体和ML特征。总体而言,在大多数肿瘤中,发现了多种TNBC类型的亚型(图3g)。因此,虽然TNBCs内最普遍的恶性群体对应于具有增殖性管腔祖细胞特征的细胞,但我们在每个肿瘤内发现了不同的细胞亚群,表明存在具有离散上皮分化状态和不同恶性转录表型的细胞。这些发现与胶质母细胞瘤的单细胞分析数据相似,显示了肿瘤内基因表达亚型的异质性。
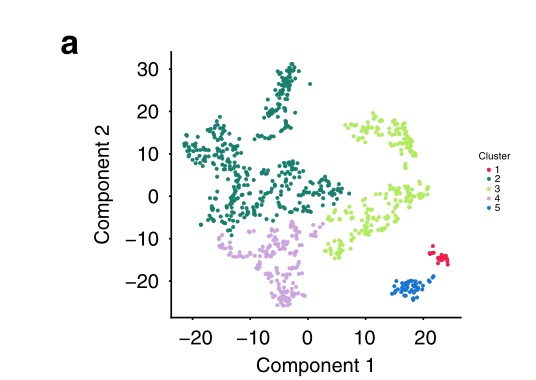
a 上皮细胞的t-SNE图,显示了五个确定的集群。通过线性回归分析排除了患者的特异性影响。
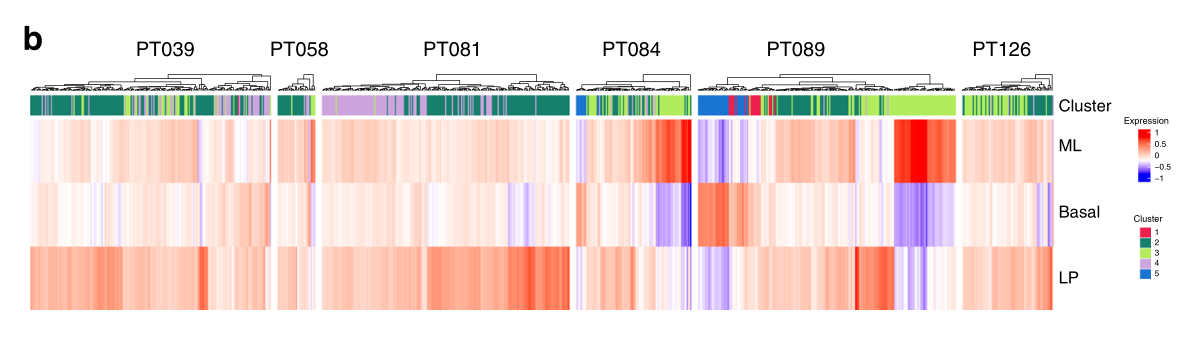
b 热图描述了分配给每个细胞的簇(顶部)和三种正常乳腺上皮亚型特征的相应表达。ML(成熟管腔)、基底和LP(管腔祖细胞)。

c 上皮细胞群中三种正常乳腺上皮亚型特征的平均表达。群组2和4最强烈表达LP特征,而群组3最高度表达ML特征。
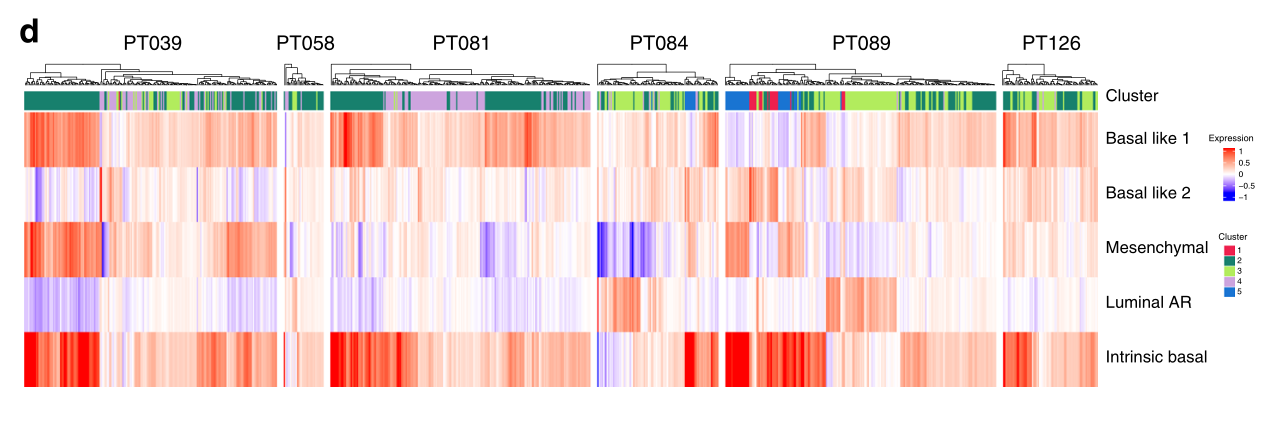
d 热图描述了分配给每个细胞的簇(顶部)和四个TNBCtype-4亚型特征和内在基底特征的相应表达。
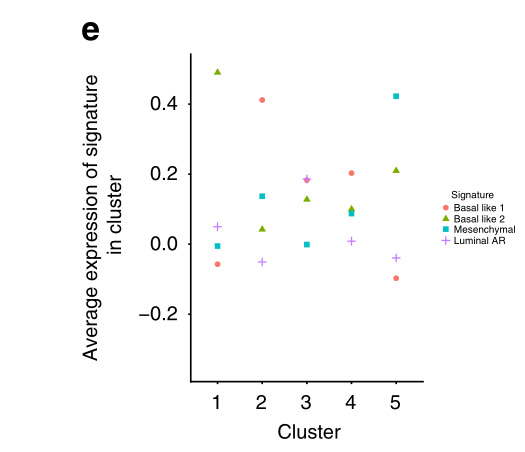
e 四个TNBCtype-4亚型特征在上皮细胞群中的平均表达量。群组2和4最强烈地表达增殖型基底样1特征,而群组3突出地表达该特征和管状AR特征。

f 将每个TNBC上皮细胞分配到单一的正常乳腺上皮亚型特征中,这取决于它对表征该特征的上调基因的平均表达量与下调基因的平均表达量之间的差异。所有肿瘤中的多数细胞都是LP型的,除了84号肿瘤,它主要是由ML型细胞组成的。
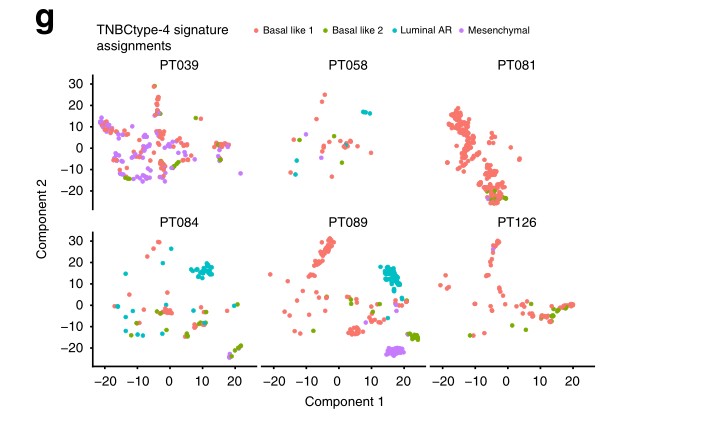
g 将每个TNBC上皮细胞分配到一个单一的TNBCtype-4亚型特征中,这取决于它的上调基因的平均表达量与下调基因的平均表达量之间的差异。每个肿瘤的细胞中都有多种TNBCtype-4亚型的表达。
5 TNBC亚群产生临床相关的特征
由于单细胞分析可以提供更强的能力来揭示导致不良临床结果的肿瘤细胞亚群,我们分析了恶性聚类亚群,以富集与侵袭性临床行为相关的独特基因表达特征(图4a)。其中包括70个基因的预后特征,该特征最初来源于对转移性复发患者与未发生转移性复发患者的原发肿瘤之间基因差异表达的分析。第二个特征(49基因转移负担特征)区分了患者来源的TNBC32小鼠异种移植模型中鉴定的单个循环转移细胞所赋予的高转移负担和低转移负担。第三个特征(354个基因残留肿瘤特征)是从接受术前化疗治疗原发性乳腺癌患者的残留存活肿瘤群体中富集的基因中获得的。值得注意的是,这些特征之间的重叠程度很小,没有一个基因出现在所有三个特征中。值得注意的是,尽管它们有不同的派生和小程度的重叠,但所有三个侵略性疾病的特征在第2群亚群中都是高度富集的(图4a,b)。
为了进一步研究第2组细胞的生物学特性,我们确定了该组细胞与所有其他上皮细胞之间差异表达的基因子集。我们发现这些第2群的选择性基因与WES分析发现的三个肿瘤的基因组拷贝数增加明显相关(39、81和84)。相比之下,其他两个恶性集群(3和4)的基因集没有显示其基因组和转录组特征之间的明显关联。这些发现共同表明,第2组细胞可能会推动肿瘤的发展,从而使那些肿瘤中含有大量此类细胞的患者获得不良的结果。
为了验证这一假设,我们接下来得出了一个独特的特征,包括第2组中差异表达最明显的基因。我们将这一特征应用于一个公开的大型数据集,其中包含了与长期患者结局相关的原发性TNBC的大量RNA-seq谱,即METABRIC队列。我们观察到第2组特征高表达的肿瘤与总生存期缩短之间存在统计学上的显著联系(图4c)。相比之下,在这个患者队列中,富集于第2群的三个原始侵袭性疾病特征本身并不能预测结果(图4b,c)。此外,定义集群1、3、4或5的特征都与该队列的临床结果无关。同样,在这个队列中,内在的基底型TNBC特征与临床结果没有关系。总的来说,这些发现揭示了单细胞分析的能力,以揭示临床相关的细胞状态,而这些细胞状态并没有通过大量的肿瘤分析被发现。
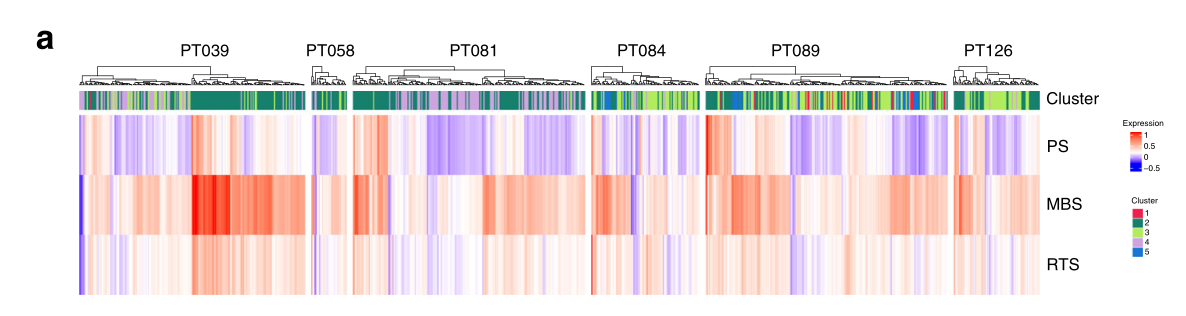
a 热图描述了分配给每个细胞的簇(顶部)和与侵袭性疾病行为相关的三个特征的相应表达:70个基因的预后签名(PS),49个基因的转移负担签名(MBS),和354个基因的残留肿瘤签名(RTS)(行)。
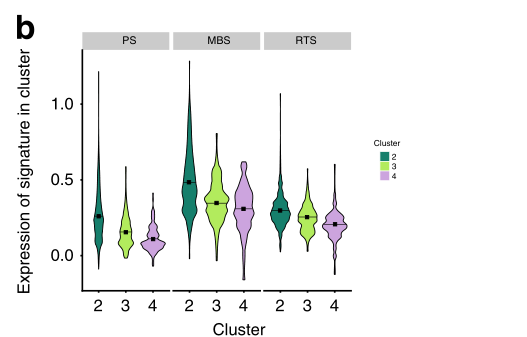
b 小提琴曲线图,表示与侵袭性疾病行为相关的三个特征的指示簇内细胞之间的表达分布。黑色方块表示每个特征在相应簇的单元中的平均表达。
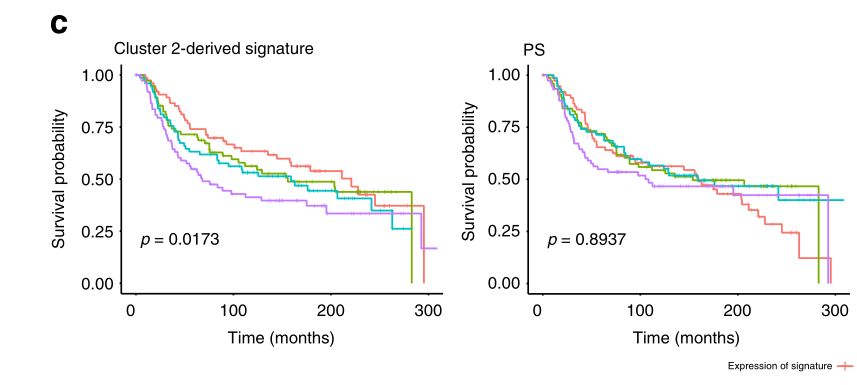

METABRIC队列中TNBC患者的Kaplan-Meier生存曲线,通过第2组衍生基因特征的表达四分位(左)。特征的高表达与较差的患者预后显著相关(对数等级检验,p=0.0173)。与攻击性疾病行为相关的其他三个特征并不能预测存活率(右)。四分位数的分离是为了可视化目的。
6 代谢和免疫程序是预后不良亚群的特征
然后,我们试图通过研究在簇2中与其他上皮细胞相比差异表达的基因中丰富的途径来确定簇2亚群背后的功能程序。我们发现,最丰富的途径与神经鞘糖脂的生物合成和溶酶体周转有关,这影响了先天性免疫系统的细胞因子途径,这些途径也是丰富的(图4d)。糖神经节苷脂最近被认为是乳腺癌中许多促进肿瘤特性的介质,包括改变的生长因子信号、EMT和干样行为。该途径中的多个关键基因在簇2亚群中选择性表达,包括糖脂转移蛋白基因GLTP和关键的鞘磷脂生物合成亚单位基因SPTLC1。鞘糖脂也被认为是先天免疫系统和获得性免疫系统的重要调节剂,并与多种上皮组织中炎症相关的癌变有关。事实上,IHC染色证实了SPTLC1在所有六个TNBC的一组细胞中的表达,此外,我们还观察到高水平的鞘氨醇-1-磷酸受体S1PR1,它在所有肿瘤的IHC染色后在一个相互反馈环中发挥作用,激活多个癌症背景下的STAT3 。另一个值得注意的与上皮天然免疫相关的簇2选择基因是GPI/AMF(葡萄糖-6磷酸异构酶/自分泌运动因子),这是一种肿瘤分泌的细胞因子,与EMT、迁移和转移有关(图4d)。此外,上皮紧密连接组装因子基因F11R的表达与乳腺癌的进展和患者的生存有关。解除对F11R等屏障因子的调控可以诱导已建立的促肿瘤细胞因子,事实上,包括CCL20和CCL22在内的这些细胞因子的子集在簇2亚群中高度选择性地表达。最后,我们确定了神经鞘糖脂途径表达本身对临床结果的影响,以验证其与簇2亚群的特异性和总体上的TNBC的相关性。我们发现,代表鞘糖脂代谢的基因特征可以预测METABRIC队列中TNBC患者的总生存期,随着表达的逐渐升高,该TNBC队列中的总生存期越来越差。总而言之,这些发现揭示了一个意想不到的TNBC细胞亚群,其转录组反映了基因组的进化,其独特的生物学特性给TNBC患者带来了糟糕的临床结果。

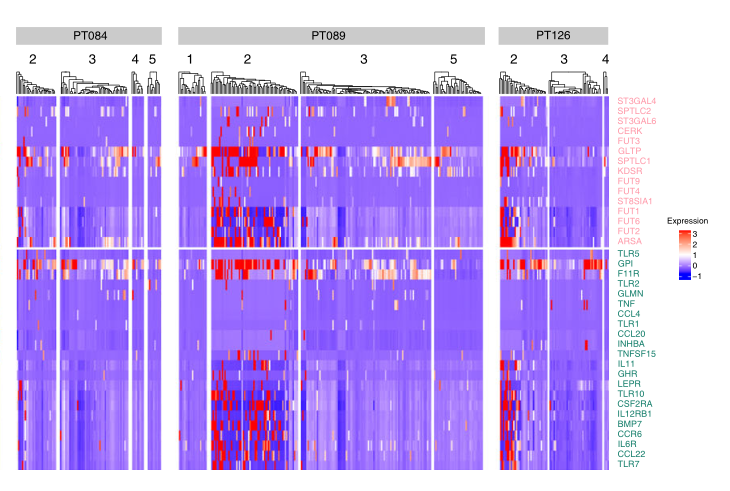
d 热图显示了所有患者的神经鞘糖脂代谢和上皮团(如上图所示)中天然免疫途径的基因表达。簇2显著丰富了这两个途径中的基因表达
四.结论
这项分析揭示了TNBC的功能异质性及其与基因组进化的关系,并发现了决定这种疾病不良结局的未曾预料到的生物学原理
五.实验流程
1 获取单细胞数据
对6个原代TNBC的>1500个细胞进行了单细胞RNA测序(scRNA-seq)。
2 数据处理
1 质控
一般来说,scRNA-seq数据预计比bulk RNA-seq数据噪音更大,主要是因为在单个细胞中可靠地转换和扩增低数量的RNA(也称为RNA捕获)是一项挑战。因此,缺失事件在scRNA-seq数据中很常见,即在多个细胞的基因中没有表达或低表达水平的情况,不一定反映真实的生物信号。不去除低质量的细胞或表达太低的基因可能会使下游分析产生偏差,使人们更难将生物信号与技术或生物噪音分开。
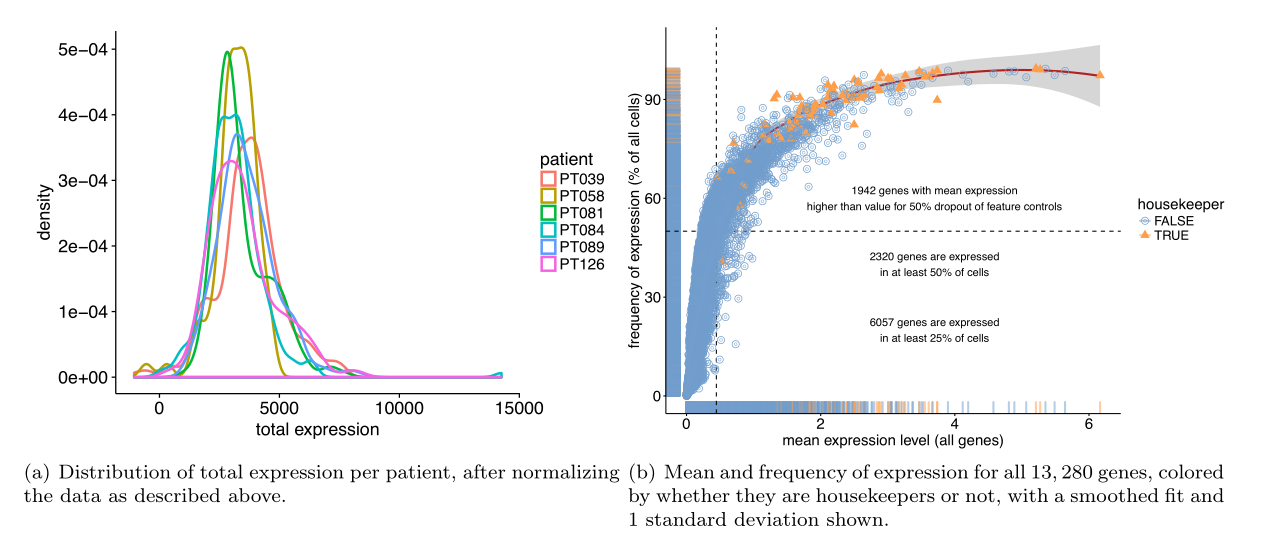
该数据集最初由6名TNBC患者的1,529个单细胞和7个样本组成,每个样本100个细胞。我们使用以下度量来识别低质量单元(如下图):
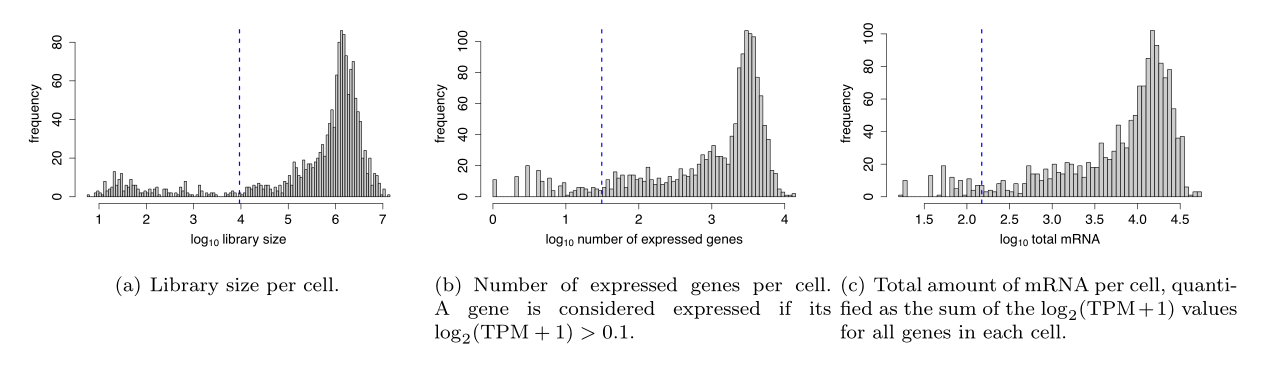
1. 文库大小:文库大小是指每个细胞的映射读数的数量。随着细胞被测序的深入,表达量的量化也更加精确。文库大小相对较小的细胞一般表明RNA在文库制备过程中没有被有效地捕获。
2. 表达基因的数量:表达基因非常少的细胞是真实细胞转录多样性的不良指标。如果一个基因的log2(TPM+1)>0.1,则认为该基因已表达。
3. mRNA总量:mRNA总量被量化为每个细胞中所有基因的log2(TPM+1)值之和。mRNA量过低的细胞表示捕获不成功,或者死亡或受损的细胞。
去除未表达的基因
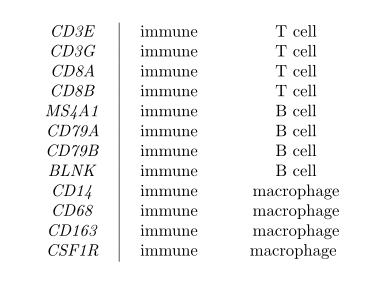
对于6个病人中的每一个,我们确定在该病人的至少95%的细胞中不表达的基因(log2(TPM + 1) < 0.1),并删除这6组的交集。要求被标记为不表达的基因在每个病人中都不表达,而不是只在所有病人中表达,这增加了我们保留与病人间异质性有关的基因的机会。在最初的21,785个基因中,过滤后还剩下13,280个。
2 标准化
归一化是预处理scRNA-seq数据的重要步骤,特别是考虑到各种混杂因素对单细胞中量化表达水平的巨大影响。混杂因素的例子包括辍学事件的频率、单细胞中mRNA的数量少、不同类型细胞的捕获之间的高变异性或技术噪音。我们的规范化策略包括以下三个步骤。
1. 用Census算法(Qiu等人,2017)将TPM值转化为相对计数(R软件包monocle(Qiu等人,2017)中的函数relative2abs)。Census通过将每个细胞中的TPM值除以每个细胞中mRNA分子的估计总数来重新衡量。通过这种方式,TPM值被转化为相对的Census计数,它是负二项式分布。这些计数已被证明(Qiu等人,2017),与单独的TPM或用其他方法归一化的计数相比,如edgeR(Robinson等人,2010)、DESeq2(Love等人,2014)或SCDE(Kharchenko等人,2014),更能反映单细胞中mRNA的实际数量。
2. scran计算特定细胞的缩放因子(也称为大小因子,即模拟滋扰性不同的随机变量),同时特别考虑scRNA-seq的高辍学率。将每个细胞的表达量除以其大小系数,是为了减少滋扰性的差异(Vallejos等,2017)。所得值在加入常数集后进一步进行对数转换。137个细胞的特定大小系数估计为0,被认为是对应于转录组多样性太低的细胞。这些细胞被从数据集中删除,剩下1189个单细胞用于下游分析(如下图)。
3. 用RUVSeq(Risso等人,2014)(RUVg)去除scran归一化普查计数中不需要的额外变异来源。RUVg使用被认为在不同样本中具有近似恒定表达的看家基因,并使用广义线性模型回归出从看家基因的表达中估计的变异。RUVg要求输入要去除的变异源的数量(k)。这里,由于数据已经在前两个步骤中被归一化,我们设定k=1。我们使用(Tirosh等人,2016)中汇编的98个管家基因的策划列表,其中93个基因存在于我们的数据集中(如下图)。请注意,这个归一化步骤可能导致负的表达值。
3 识别细胞类型。
我们采用了两步组合的方法来确定肿瘤所包括的不同细胞类型。(1)基于文献的特定表达标志物清单,以前建立了定义细胞类型;(2)聚类
表达标志物
用于分类细胞类型的基于文献的列表由49个表达标记物组成,专门针对四种细胞类型,由多个参考文献汇编而成(Tirosh等人,2016)(表7)。为了尽量减少错误分配的数量,我们只在认为有足够的基于表达的证据支持该分配时才将细胞分配到特定的细胞类型(表达阈值=1)。

在这第一步之后,未被归入上述任何类别的细胞被标记为未知,而属于上述一个以上类别的细胞被标记为未决定。特殊情况下,同时表达上皮和基质标志物的细胞被认为表示上皮-间质转化(EMT )状态,并被分配到上皮类。除了表7中的标记,我们还使用(Tirosh等人,2016)中建议的标记,测试任何单细胞是否是自然杀伤细胞或树突状细胞,然而在我们的数据集中没有发现任何此类细胞。在这个分类步骤之后,每个细胞类型的结果分布显示在表8中。
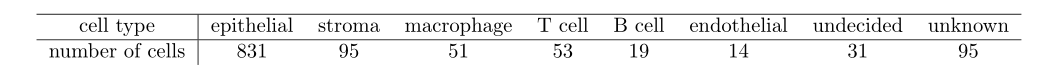
聚类
然而,仅根据表达标记对细胞进行分类可能是不精确的,特别是考虑到scRNA-seq数据的高辍学率。因此,我们接下来将基于t-SNE的聚类应用于细胞向低维空间的投影,这在Monocle(Qiu等人,2017)中实现,并受Seurat(Butler和Satija,2017)启发。在聚类之前,我们回归出病人的e↵ect,以使产生的聚类不主要是36个代表不同的病人。此外,我们选择了一个表达基因的子集(平均表达量>0.1),在细胞间具有较高的分散性,以增加推断出的集群的稳健性,如(Qiu等人,2017)所讨论的。通过首先计算经验分散值,然后拟合分散-平均关系,为每个基因选择一个分散参数,如DESeq中的 estimateDispersions(Love等人,2014)所实现的。所选基因的经验离散度高于或等于离散度拟合值。聚类的数量是由Monocle自动选择的,实现了(Rodriguez and Laio, 2014)中介绍的基于密度的方法。
我们在1,189个细胞的数据集中确定了9个聚类。鉴于每个簇中细胞的高度相似性,我们通过尝试分配未知和未决定的细胞,以及纠正以前基于标记的分配来完善每个簇。具体来说,对于每个集群,如果至少80%的分配细胞是单一类型的,我们计算该集群中的未知和未决定的细胞对每个标记类的平均表达。如果未分配的细胞对该簇的流行细胞类型显示出最高的平均标记物表达,那么我们就分配它们。此外,由于这些集群由一种流行的细胞类型组成,我们评估同一集群中其他细胞类型的平均标记物表达量是否为流行的细胞类型最高。在这种情况下,我们将这些细胞重新分配给流行的细胞类型。另外,对于不包含单一优势细胞类型的簇(<80%的分配细胞),我们既不分配任何未知或未识别的细胞,也不重新分配任何细胞。
在这两个分类步骤之后,仍有19个细胞属于未决定的细胞类型,58个细胞仍然是未知的。我们将这77个细胞从我们的数据集中删除,现在由1,112个细胞组成。由此产生的细胞类型分布如表9所示。

4 识别循环细胞
在识别循环细胞时,我们遵循了(Tirosh等人,2016)中描述的框架,其中细胞周期的G1/S和G2/M阶段的分数是通过平均一组相关基因的表达来计算的。
在43个代表G1/S期的基因中,有41个也在我们的数据集中被确认,而在55个代表G2/M期的基因中,我们在数据集中确认了49个。循环细胞被定义为具有高G1/S分数或G2/M分数的细胞,而非循环细胞是具有低G1/S和G2/M分数的细胞。与(Tirosh等人,2016)的分析不同,该分析使用固定的阈值来决定一个分数是高还是低,我们使用数据得出的阈值,即每个分数高于中位数2MAD。这相当于将214个细胞归类为周期性,其余898个归类为非周期性。
5 从scRNA-seq数据中识别拷贝数的改变
我们通过从每个细胞的表达中减去一项不同研究(Gao等人,2017)中剖析的240个正常上皮细胞的平均表达,来规范我们的单细胞的表达谱。20,337个转录物在我们的数据集和分析正常上皮细胞的数据集之间是共同的。我们遵循其他地方描述的这种特殊分析的预处理步骤(Tirosh等人,2016;Patel等人,2014;Gao等人,2017),即我们将表达量量化为log2(TPM + 1)/10,并删除所有细胞中平均表达量<0.1的所有基因。这相当于保留了4,673个转录本。我们根据基因的染色体位置对其进行排序,并删除240个正常细胞的平均表达。我们进一步对所有大于3和小于㼿3的表达进行分级。我们将每个基因的拷贝数值定义为窗口大小为100,以每个基因为中心的滑动平均值。最后,对于每个基因,我们将得到的拷贝数值在所有细胞中居中。
6 上皮细胞的聚类
我们对868个上皮细胞进行聚类,其方式与聚类小节中描述的类似,即采用Monocle中开发的算法,并对患者的差异进行退步处理。聚类的数量是由Monocle自动选择的,实现了(Rodriguez和Laio, 2014)中介绍的基于密度的方法。我们确定了五个上皮细胞集群,如下:集群1:22个细胞,集群2:398个细胞,集群3:231个细胞,集群4:170个细胞,集群5:47个细胞。
7 基因表达特征
我们用下调基因的平均表达量减去上调基因的平均表达量,计算出每个细胞在三个正常乳腺特征(Lim et al, 2009a)下的表达量。我们将每个细胞分配到其表达量最高的特征中。这三个正常的乳腺特征是:
成熟管腔细胞(ML):561个上调基因和257个下调基因,其中384个上调基因和197个下调基因也在我们的数据中;
基质细胞:942个上调基因和942个下调基因,其中588个上调基因和747个下调基因也在我们的数据中;
管腔祖细胞(LP):358个上调基因和179个下调基因,其中231个上调基因和139个下调基因也在我们的数据中。
类似地,我们通过从上调基因的平均表达中减去下调基因的平均表达来计算四个TNBCtype4特征(Lehmann等人,2016)下每个细胞的表达,并将每个细胞分配给它表达最高的签名。TNBCtype4特征是:
basal like 1:320个上调基因和358个下调基因,其中272个上调基因和291个下调基因也在我们的数据中;
basal like 2:222个上调基因和217个下调基因,其中197个上调基因和165个下调基因也在我们的数据中;
mesenchymal:314个上调基因和542个下调基因,其中283个上调基因和448个下调基因也在我们的数据中;
Lumina AR:1093个上调基因和1143个下调基因,其中923个上调基因和987个下调基因也在我们的数据中。
8 生存分析
我们对2017年11月20日从cBio门户网站(Gao等人,2013年)下载的METABRIC mRNA数据集(Pereira等人,2016)进行生存分析,包括299名TNBC患者。拟合出COX比例风险回归模型,p值由对数秩检验法得到。
9 与群集2相关的特征
我们使用在Monocle中实现的功能差异基因测试来识别在簇2中差异表达的基因,并与所有其他上皮细胞进行比较。3262个基因以0.1FDR值差异表达。我们评估了前10个差异表达基因:GLTP、Skp2、Hp、PGAM5、Gp1、NUDT19、INIP、ECT2、S1PR2、MS4A10,其中9个(除MS4A10外)在METABRIC数据集中。我们使用特征中9个基因的表达总和作为预测因子,并根据表达总和的分布,按患者数量的四分位数对生存数据进行分层。
10 糖磷脂代谢
通过查询人类基因数据库GeneCard中的鞘磷脂代谢,获得了105个糖脂代谢途径的基因成员(补充表4)。其中102个基因(除STS、GM2A、GDF1外)在METABRIC数据库中。我们使用通路中102个基因的表达总和作为预测因子,并为了可视化的目的,根据表达总和的分布,按患者数量的三分位数对生存数据进行分层。
六.附录
缩写
三阴性乳腺癌(TNBC)
雌激素受体(ER)
孕激素受体(PGR)
人表皮生长因子受体2(HER2)
下一代测序(NGS)
拷贝数变异(CNV)
全外显子组测序(WES)