近年来,随着高通量测序技术的发展,转录组测序已经成为研究基因表达调控的主要手段。我们知道,很多物种的转录本非常多样和复杂,绝大多数真核生物基因不符合“一基因一转录本”的模式,这些基因往往存在多种剪切形式。通过二代测序,可以很准确地进行基因的表达及定量的研究,但是由于读长的限制,不能得到全长转录本的信息。因此,基于三代测序平台的全长转录组成为新的研究热潮。
全长转录组(Full-length transcriptome)是基于PacBio和Nanopore三代测序平台,无需打断拼接,直接获得包含5’UTR、3’UTR、polyA尾的mRNA全长序列及完整结构信息,从而准确分析有参考基因组物种可变剪接及融合基因等结构信息,克服无参考基因组物种转录本拼接较短、信息不完整的难题。同时还可以借助二代测序数据,进行转录本特异性表达分析,获得更加全面的注释信息。
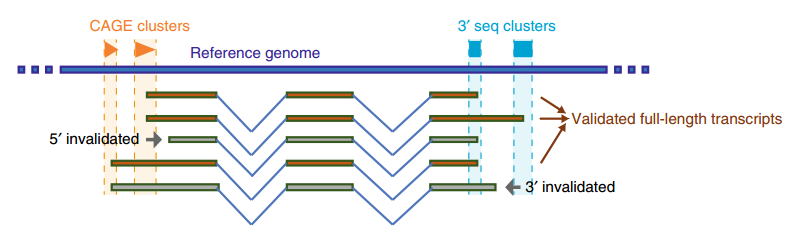
图1 大鼠中已证实的全长转录本
全长转录组测序原理
实验流程
ShiYanLiuCh

图2 全长转录组实验流程
CCS测序(滚环测序)
GunHuanCeXu
全长转录组采用单分子实时测序技术,通过构建哑铃型文库,以环形方式循环测序。测序时,短片段文库更容易落入零模波导孔(ZMWs)。
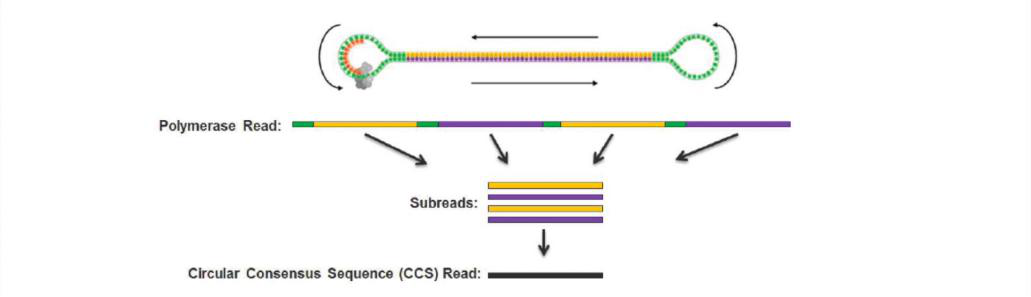
图3 全长转录组CCS测序
-
Polymerase read: 三代测序直接读取的序列,包含接头和插入片段;
-
Subreads: 将Polymerase read接头去除,插入片段每测一次,生成一条subread;
-
CCS read: 来源于同一条Polymerase read的subreads (至少两条)生成的一致性序列。
全长转录项目设计
针对有参考基因组的物种,全长转录组信息可以纠正基因组的错误组装、更准确地发现新的转录本和基因、分析基因融合事件等。
针对参考基因组不完善的物种,全长转录组可优化基因结构,辅助基因组组装和注释,提高基因表达量准确性和数据利用率。
针对无参考基因组的物种,通过三代全长转录组测序来构建物种Unigene库,无需进行序列组装,就可以获得该物种转录组水平的参考序列(转录组水平的参考基因组),为后续研究提供很好的遗传信息基础。
转录组具有时空特异性,根据研究目的进行实验设计,如下:
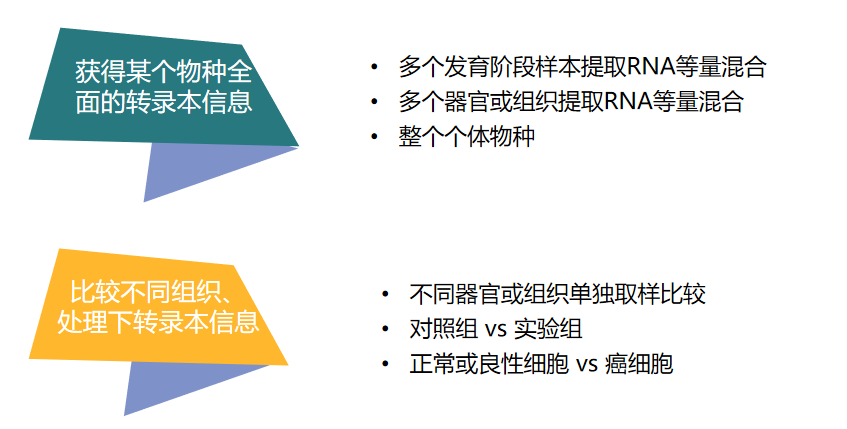
图4 全长转录组实验设计

建议2+3代转录组测序技术同时使用,保证结构准确性、序列完整性及序列表达量准确性,达到数据的最优利用效果以及性价比最高。针对不同要求及目的,对于大部分物种来说推荐测序数据量8-12G,研究低表达基因、可变剪切事件、多倍体或者基因数目较多的物种,需适量增加数据量。
全长转录组分析流程
获得原始测序序列(Sequenced Reads)后,通过如下流程进行生物信息分析:
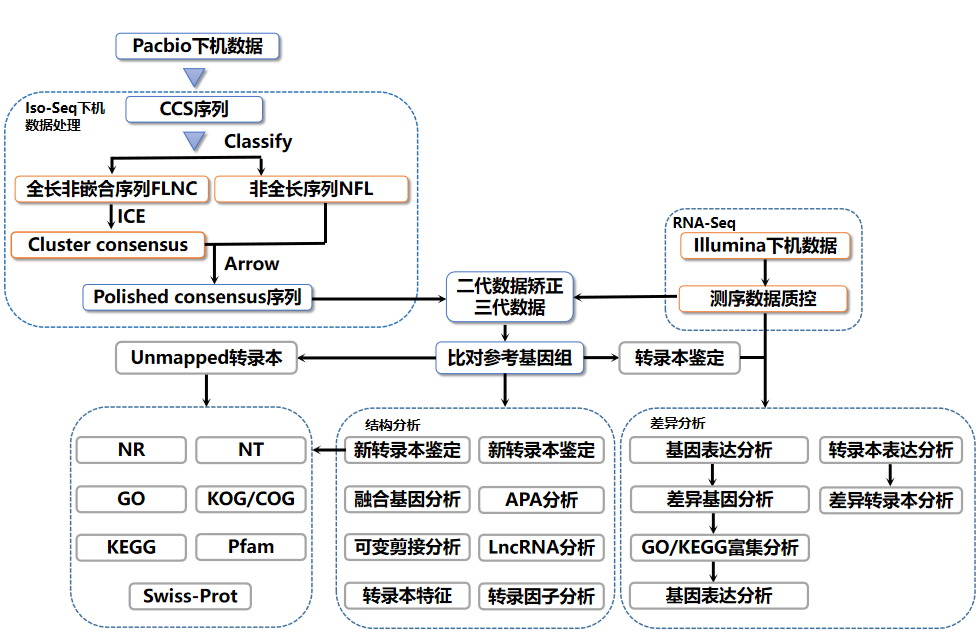
图5 凌恩生物全长转录组生物信息分析流程
凌恩生物全长转录组测序优势
-
无需进行转录本拼接,能够直接获得全长转录本序列,从而更真实的反映测序物种的转录组信息;
-
能够检测多种可变剪切形式,发现更多的剪接位点和可变剪切事件;
-
能够发现新的功能基因,补充基因组注释;
-
有助于融合基因、同源基因、超家族基因或等位基因的精确分析;
-
专业的生信分析团队,项目经验丰富。
送样要求
-
RNA样品总量要求
一般总RNA>5 ug/文库,总量>15 ug(3个文库),浓度≧250ng/ul。
-
RNA样品质量要求
OD260/280≧1.8,OD260/230≧1.0,260nm处有正常峰值;总RNA的RIN值≧8.0,28S/18S≧1.3。
-
避免cDNA损伤
采用安全染料,避免EB和紫外照射对cDNA造成损伤。
总之,三代全长转录组对于有参物种和无参物种均适用,获得更多的转录调控事件,从而更真实的反映测序物种的转录组信息。
目前,全长转录组测序研究基因结构已经成为发文章的趋势,研究物种包括高粱、玉米、拟南芥、鸡、人和小鼠、毛竹、棉花等。凌恩生物提供专业的全长转录组测序及分析服务,助力您的研究!

用心做好测序,交托每一分成果