Nature Reviews Genetics| 在基因组学中应用机器学习的常见陷阱
Whalen, S., Schreiber, J., Noble, W.S. et al. Navigating the pitfalls of applying machine learning in genomics. Nat Rev Genet (2021). https://doi.org/10.1038/s41576-021-00434-9
Tips:本文内容较干,可以点赞、在看、收藏后在PC端细品,且PC端排版更佳。
如今,遗传(genetic)、表观基因组学(epigenomic)、转录组学(transcriptomic)、化学信息学(cheminformatic)和蛋白质组学(proteomic)数据的规模,加上易于使用的机器学习(ML)工具包,推动了监督学习在基因组学研究中的应用。
然而,ML软件中 统计模型(statistical models) 和 性能评估(performance evaluations) 背后的假设在 生物系统(biological systems) 中常常不满足。
在这篇综述中,作者举例说明了在基因组学中应用监督式(supervised) 机器学习(ML)时遇到的几个常见陷阱(pitfalls)的影响。探索了基因组数据结构(the structure ofgenomics data)如何影响性能评估和预测。
监督式ML(supervised ML),其目标是学习一个模型,通过识别预测模型下的基因组特征,对新数据进行准确预测和/或产生机械洞察力(mechanistic insight)。
作者本文中所用的所有示例数据及代码都上传在:https://github.com/shwhalen/ml-pitfalls,可自行下载。或者点赞、在看本篇推文,截图发我微信
mzbj0001进行领取。

性能评估的术语
Cross- validation (交叉验证)
例如,10折交叉验证(10-fold cross validation),将数据集分成十份,轮流将其中9份做训练,1份做验证,10次的结果的均值作为对算法精度的估计,一般还需要进行多次10折交叉验证求均值,例如:10次10折交叉验证,以求更精确一点。
Performance statistics (性能评估指标)
真阳率(True Positive Rate, TPR)
检测出来的真阳性样本数除以所有真实阳性样本数。
假阳率(False Positive Rate, FPR)
检测出来的假阳性样本数除以所有真实阴性样本数。
精确率(Precision)
真阳性的数目除以预测阳性的总数(真阳性加上假阳性)。
错误发现比例(False discovery proportion,FDP)、错误发现率(False discovery rate,FDR)
FDP 为 1−Precision。在多次实验中,预期的(或平均的) FDP 是 FDR。
Accuracy
准确率是分类器做出的正确预测的比例。它的计算方法是,正确预测的数量除以预测的总数。
Visualizing performance (性能可视化)
Receiver operating characteristic (ROC) curve
受试者工作特征曲线(ROC)描绘出真实阳性率与FPR的函数关系。该曲线下面积(auROC)量化了分类器的性能,面积为1.0代表完美性能,面积为0.5代表随机机会。auROC 不依赖于测试集中的类别比例。
Precision–Recall (PR) curve
准确度-召回率(PR)曲线将准确度作为召回率(也称为真实阳性率)的函数绘制出来。完美性能对应的面积(auPR)为1.0,而随机机会对应的面积为 阳性个数/示例总数。对于一个固定分类器,auPR 将根据测试集中的类别比例变化。
陷阱1: distributional differences(分布差异)
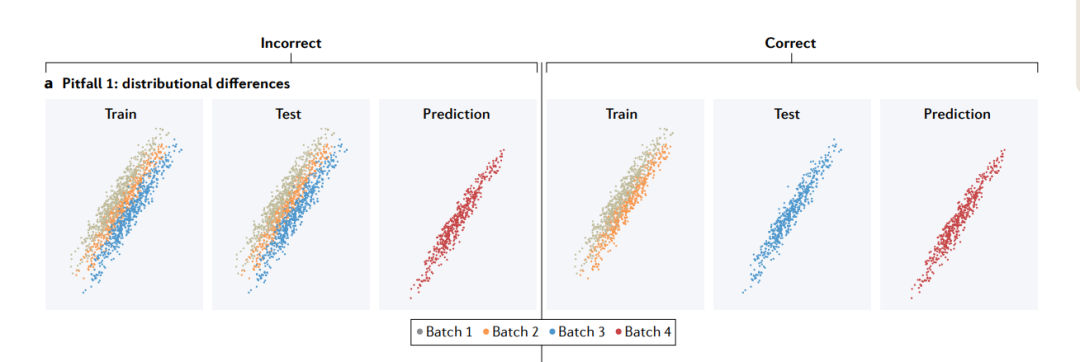
分布上的差异可能来自不同的来源,例如批次效应。如果训练集和测试集混合了来自不同批次的示例(左),测试集的性能将比新批的性能高得多。为了拟合适用于新批的模型,训练集和测试集应该由不同的批次组成(右)。
分布差异会影响特征(features)(其边际分布(marginal distribution)表示为P(x))、结果(outcome )(P(y))和/或特征与结果之间的关系(条件分布(conditional distribution)P(y|x))。
在基因组学(genomics)中,由于许多原因产生了分布差异。一个常见的原因是数据中固有的生物结构(inherent biological structure)。例如,常染色质(euchromatin)和异染色质(heterochromatin)之间的表观遗传谱(epigenetic profiles)不同。蛋白质属于功能类别,每一个都有不同的表达模式和物理相互作用。在全基因组关联研究(GWAS)中,当变异发现或模型训练的个体与不同的祖先(ancestry)的人群进行基因型-表型关联(genotype–phenotype associations)测试时,群体遗传结构产生了分布差异,称为确认偏倚(ascertainment bias)。当一个模型在不同的生物环境(biological contexts)(例如,不同的细胞类型、不同的物种或在体外或体内)中训练和应用时,也会产生分布差异。最后,由于研究设计和技术因素,分布可能会有所不同。批次效应(batch effects)在基因组学中非常普遍,并且会使测量值的平均值和变异性产生偏差。
Ascertainment bias arises when data for a study or an analysis are collected (or surveyed, screened, or recorded) such that some members of the target population are less likely to be included in the final results than others. Ascertainment bias is related to sampling bias, selection bias, detection bias, and observer bias.
Ascertainment bias can happen when there is more intense surveillance or screening for outcomes among exposed individuals than among unexposed individuals, or differential recording of outcomes.
Catalogue of Biases Collaboration, Spencer EA, Brassey J. Ascertainment bias. In: Catalogue Of Bias 2017: https://catalogofbias.org/biases/ascertainment-bias/
在这个陷阱中,训练集和测试集有相同的分布,而预测集有不同的分布。在这种情况下,应该期望交叉验证的性能比预测集的性能更高。由于预测集和测试集在分布上存在差异,在模型拟合过程中学习到的特征和结果之间的关系在预测中可能不成立。
幸运的是,一些分布上的差异很容易识别。理想情况下,应该检查结果和特征的边际分布。但是在预测设置中,结果通常是未知的,所以人们只能评估特征分布。可视化是一种简单的方法,可以将数据投影到两个维度并绘制散点图,也可以比较特征值的直方图。更复杂的方法是使用统计检验来检测分布的差异;例如,对二元特征的二项检验(binomial test),对单变量连续特征的Kolmogorov-Smirnov检验或对多元连续特征的最大平均偏差(Maximum Mean Discrepancy)。基于模型的离群点和异常检测技术也可以使用。
对分布差异的解释仍然是一个开放研究领域。通常使用各种批次校正方法,如分位数归一化(quantile normalization)、使用ComBat对测量变量进行经验贝叶斯调整(empirical Bayes adjustment)、用于估计和校正未知噪声源的替代变量分析(surrogate variable analysis,SVA)以及能够识别批次之间公共模式的相关性分析。另一个解决方案是对抗学习(adversarial learning),这是一组试图通过提供欺骗性输入来欺骗模型的技术。具体来说,可以使用一个训练有素的模型来预测每个示例来自的数据集,从而为主要预测任务生成惩罚(penalties)。
陷阱2: dependent examples(相关例子)
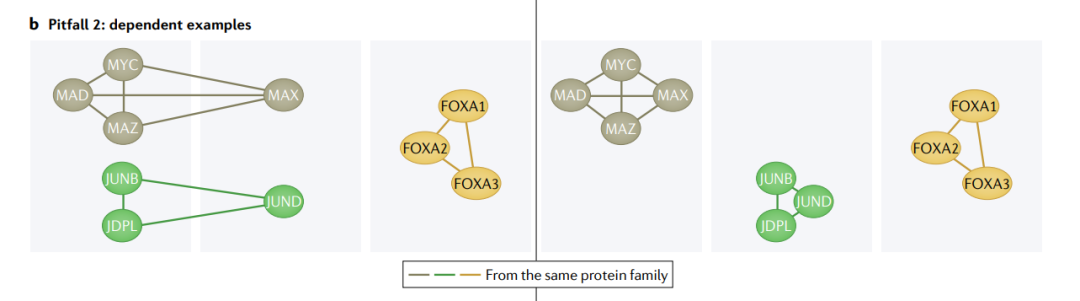
当生物群体表现出相似的特征-结果关系时,例如来自同一家族或复合体的蛋白质的相关功能,依赖性结构就出现了。如果使用同一组中的其他实体训练一个模型,预测结果就很容易(左)。为了确保模型可以泛化,我们应该将整个生物组划分为训练集或测试集(右)。
常用的ML模型和交叉验证的数学依赖于独立性假设,这意味着一个例子的值不依赖于另一个例子。例如,在不替换已抽牌的情况下从一副牌组中重复抽牌是有相关性的,因为下一张牌的概率取决于已经抽到的牌。
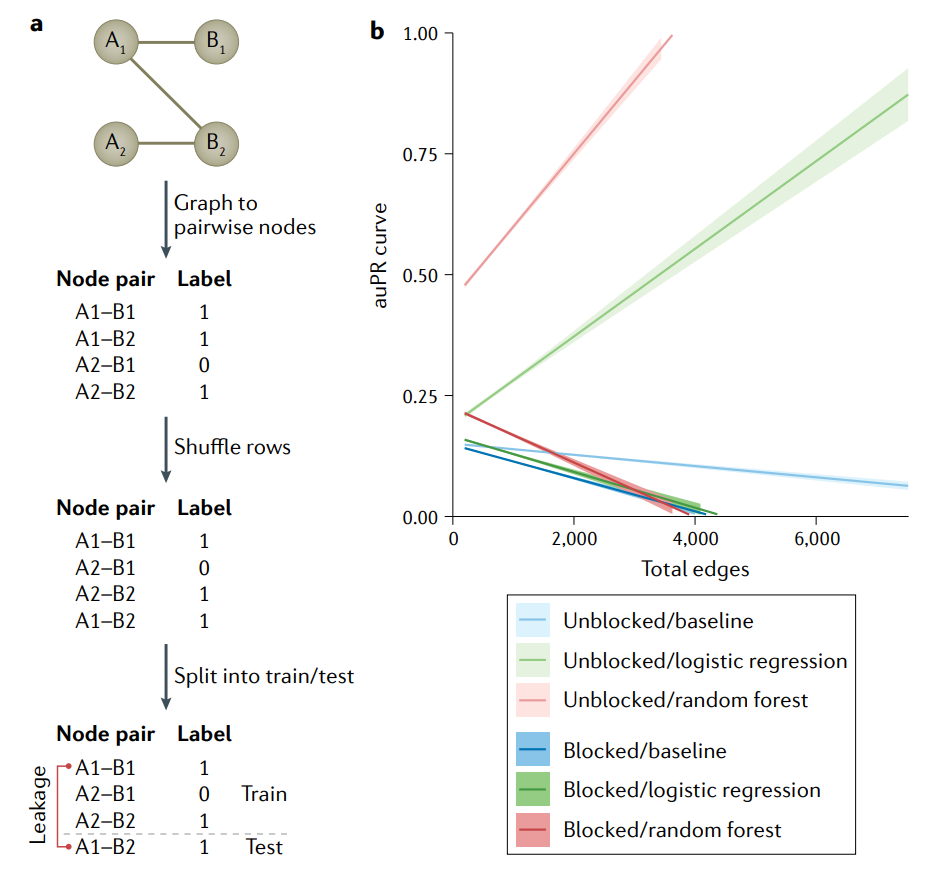
生物网络中的节点对不是独立的。
a图|对集合A、B的相互作用网络图进行特征的编码,有相互作用标注为
1,没有相互作用标注为0。得到的特征矩阵和标签可用于标准的机器学习(ML)算法。为了估计模型的性能,数据集被随机地分成训练集和测试集。节点A1的一条边碰巧在训练集中,另一条在测试集中。这些样本是相互依赖的,因此导致信息在train–test中泄漏(leak across),从而夸大了性能。这个问题已经影响到基因组学的各个领域,其中集合A和B可能是蛋白质和配体,药物和目标基因或增强子和启动子。b图|基于图(相互作用网络)的数据集的性能膨胀随着边数的增加而增长。使用Group k fold cross validation也称为blocking,防止非独立的例子跨越train–test鸿沟。可以看到未阻塞交叉验证的auPR值被错误地夸大了,原因是训练集中的节点对与测试集中的节点对之间存在依赖性,模型可以对测试集进行准确的预测。问题随着边数的增加而增加,因为有共享节点的边穿过train–test分水岭的概率增加。
在基因组学中,依赖性是普遍(pervasive)存在的,但很难识别。当预测蛋白质之间的相互作用时(protein–protein interactions),例子是蛋白质对。当配对在数据集中用唯一标识符表示时,它们可能看起来是独立的,但所有共享给定蛋白质的配对都是相互关联的。增强子-启动子(enhancer–promoter)、调节子-基因(regulator–gene)和药物-蛋白相互作用(drug–protein interactions)的相关例子也类似。
独立性和相同分布假设经常纠缠(entangled)在一起。例如,家庭成员的基因分型结果是相互依赖的,而且可能在分布上与其他家庭不同。然而,依赖关系并不总是已知的。在监督ML分析中,即使已知的依赖关系也有可能被忽略。当数据被格式化为一个表格,每行有一个示例时(大多数ML工具包的标准),很容易进行模型拟合和交叉验证,而无需检查示例是否独立。
不考虑示例之间的依赖关系可能会导致偏倚模型(biased models)和对模型性能的过度乐观估计。随机交叉验证不能防止这个问题,并高估了性能,因为测试集中的示例可以与训练示例相关联,并将不应该存在的信息带入测试集中。例如,一个预测蛋白质-蛋白质相互作用的模型在交叉验证中可能比在新蛋白质上表现得更好,因为具有不止一个相互作用的蛋白质可以出现在每个折叠的训练集和测试集上。这个问题的规模随着依赖程度的增加而增加;在高度相关(highly connected)的图和具有hub节点的图中,精度召回曲线下的面积(area under the precision–recall ,auPR)可以提高0.5以上。
有几种方法可以减轻依赖示例对ML模型的影响,而且这些方法并不相互排斥。最好的解决方法是在模型评估阶段承认依赖关系,减少过拟合。Group k fold cross validation 是一个防止非独立的例子跨越train–test鸿沟的壁垒。它不能减少依赖,也不是一个普遍的解决方案;有些依赖结构太复杂,无法用阻塞来解决。但它确实防止了由训练和测试集中的依赖示例引起的夸大性能指标,使性能更接近于独立预测集中的预期。另一种方法是通过对边下采样(downsampling edges)(例如,每个高度节点只包含一条边)或降低高度节点的权重(downweighting nodes with high degree)(例如,使用节点倾向评分)来直接降低依赖性。然而,这些策略可能使修改的数据在生物学上不现实,例如,通过删除高度交互的蛋白质或基因组区域。第三种选择是使用明确地为实例之间的协方差建模的方法,例如来自生物统计学的混合效应模型(mixed effects models)、时间序列模型以及来自空间统计的自相关模型(autocorrelation models)。然而,这些模型可能无法扩展到大型基因组数据集,而且很少使用通用的ML工具包实现。
陷阱3: confounding(混杂)

混杂变量是无法观察到的变量,它改变了被观察变量之间的依赖结构。在本例中,未测量的个体世系是遗传变异和基因表达之间关系的混杂物(左),导致C等位基因似乎与高表达相关。在对个体世系进行调整后(右),我们看到来自A4祖先人群(ancestry group)的个体表达更高。C等位基因和表达之间的联系仅仅是因为C在A4以上的个体中更常见。尽管很难发现,混杂因素应该明确地包括在建模方法和训练和测试集的构建中。
最难诊断的一个陷阱涉及数据,其中一个未测量或人工变量(artefactual)(“混杂器”)创建或掩盖了与结果的关联。这是因为混杂因素导致特征和结果之间的依赖性。误差在于混杂因素没有被测量或认为是不重要的,因此不包括在模型中。这可能很少或根本没有影响预测的准确性,但它会导致错误的解释特征-结果关系(feature–outcome relationships);以及当模型应用于没有混杂器或与原始背景(original context)分布不同的新背景中时的不良性能。
基因组数据中缺乏容易解释的线索,这意味着混杂因素很难识别。在图像分析中,当混杂物出现在我们眼前时,它们仍然很难识别。例如,背景风景(background scenery)混杂预测动物类型和放射扫描仪类型混杂预测髋部骨折。遗传研究中的混杂可能源于未建模的环境因素、种群结构以及其他因素。基因组学的一个例子是通过表观遗传学特征预测三维染色质相互作用(3D chromatin interaction)数据(例如Hi-C)。线性基因组中相互接近的一对位点具有相似的表观遗传标记,而且由于聚合物物理的原因,它们在3D中也经常相互作用,使得基因组距离(genomic distance)成为一个混杂因素。另一个常见的例子是,在不同批次抽样或处理具有不同结果(例如,患病与健康或不同治疗组)的样本,无意中混杂了数据。当整合来自多个研究的数据时,不同条件或细胞类型的基因组分析或生物信息学管道(bioinformatics pipelines)的差异也会造成混杂。因此,这意味着在任何基因组数据集中都可能存在一定程度的混杂。
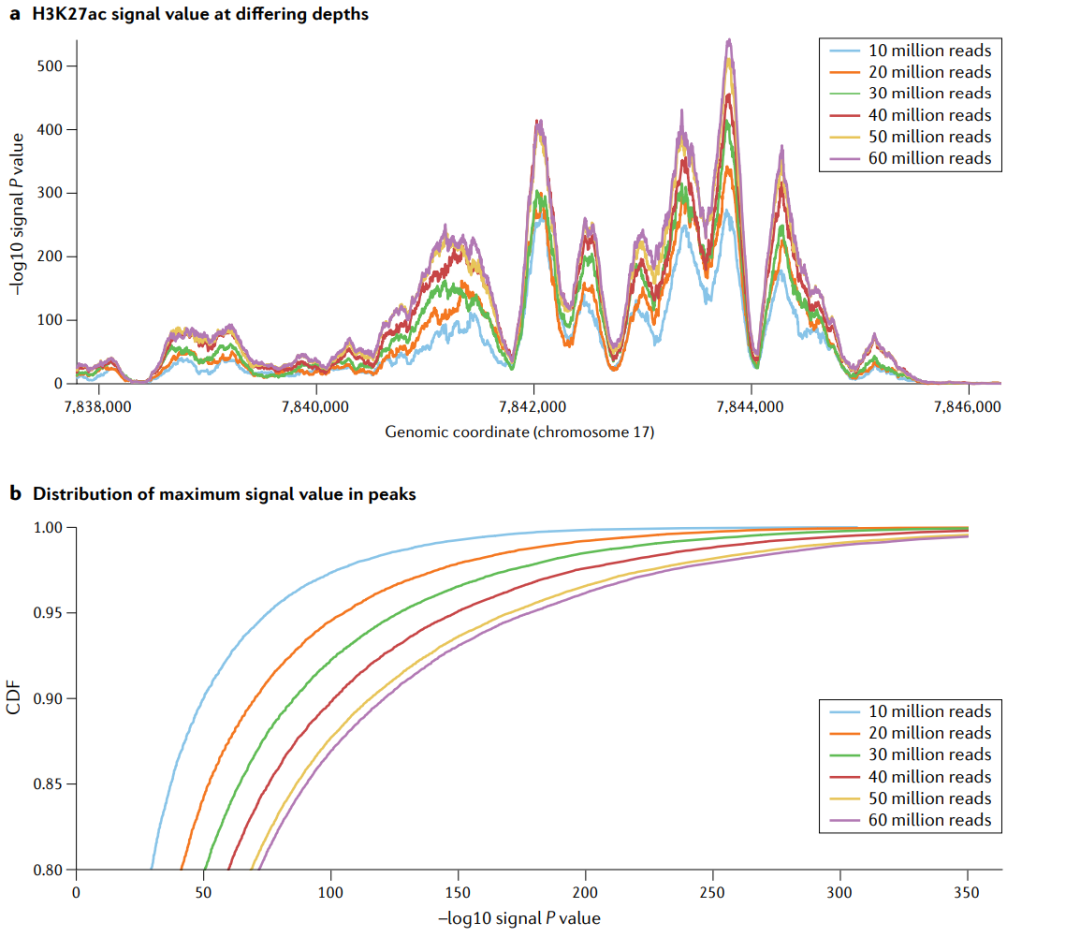
测序深度作为一个混杂变量。在平滑肌细胞中进行6个测序深度再加工的H3K27ac实验。a|不同测序深度的H3K27ac信号值。−log10p值信号位于17号染色体的一个位点上。随着序列测序深度的增加,峰值也越来越高,如果在机器学习模型中没有考虑到测序深度,那么测序深度就成为了一个混杂因素。这种趋势在一些峰值中比在另一些峰值中更大,表明混杂效应是非线性的。b|最大信号值在峰值中的分布。同样的一组全基因组峰值,即使用1000万个读取量,用于所有的测序深度。低读深度的累积分布函数(cumulative distribution function,CDF)曲线高于高读深度CDF曲线,这意味着在任何给定的P值阈值下,更少的峰值被称为显著的。机器学习模型的目标是预测峰值或学习显著峰值中丰富的特征,如果它们不考虑混杂,那么当应用于具有不同测序深度的数据时,机器学习模型将会有偏差。
这个陷阱的主要问题是,ML模型将估计结果和依赖于混杂的特征之间的关联,但建模者将错误地将其解释为直接的生物效应(direct biological effect)。问题是我们没有意识到混杂器对观察数据的影响,并且我们没有将它包含在ML模型中。重要的是,交叉验证不能防止混杂效应,因为混杂在训练集和测试集中都存在。当拟合的模型在一个新的环境中做出不准确的预测时,即混杂因素不存在或与测量的特征和结果有不同的关系时,混杂的关联通常会浮出水面(come to light)。例如,如果世系(ancestry)在一个训练队列中混杂了基因型-表型(genotype–phenotype relationships)关系,那么拟合的ML模型在应用于一个世系是随机的队列时将表现不佳。除了制造错误的关联,混杂还会掩盖真实的关系。混杂因素带来的可变性使得很难了解特征和结果之间的真实关系。
有几种统计方法有助于预防这些问题。理想情况下,对于可能的混杂因素,例如实验批次,示例应该是随机的。当这是不可能的时候,一个解决方案是使用主成分(principal components)、表达式残差的概率估计(PEER)或其他统计数据来概括高维数据中的结构来捕获未测量的混杂因素(unmeasured confounders)。也可以尝试测量潜在的混杂变量。在这两种情况下,包括在ML模型中的变量将调整其效果并减少混杂。在从表观遗传特征预测3D染色质相互作用的例子中,包括模型中的基因组距离,澄清了表观遗传标记与3D相互作用的相关性是否比预期的更强。许多回归模型总是可以包含一个变量,并据此进行调整。但是随机选择特征的ML方法很难将混杂因素强制放入模型中。为了解决这个问题,正在开发针对有监督的和无监督的ML模型的对抗性策略(adversarial strategies)。或者,一个被测量的变量可以被用作基线预测器,可以与具有其他特征的模型进行比较。需要注意的是,在模型中添加一个变量以减少混杂,当它充当碰撞器时,可能会引起偏差。或者,一个被测量的变量可以被用作基线预测器(baseline predictor),可以与具有其他特征的模型进行比较。需要注意的是,在模型中添加一个变量以减少混杂,当它充当碰撞器(collider)时,可能会引起偏差。
陷阱4: leaky preprocessing(数据预处理的泄露)
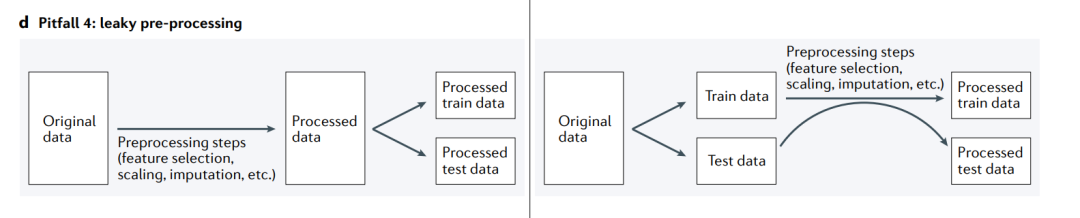
当由于训练集和测试集一起进行预处理而将信息从测试集泄漏到训练集时,就会发生信息泄漏(左)。相反,应该将原始数据分割为训练集和测试集,并分别进行预处理(右)。
在ML分析中,一个微妙但普遍存在的问题是,数据处理会不经意地导致信息从测试集泄漏到训练集。当训练集以依赖于测试集数据的方式处理时,就会发生信息泄漏(“双倾斜,double dipping”),这会导致示例之间的依赖性(陷阱2),并干扰测试集用于评估模型性能的效用。
泄漏的预处理在基因组学中很普遍。同时查看多个示例的任何数据转换都可能存在问题。具体的方法包括标准化和主成分分析(PCA),加上各种其他缩放和无监督嵌入方法(unsupervised embedding approaches)。监督特征选择,包括过滤基于结果关联的特征,是另一种形式的预处理,当在交叉验证之外执行时可能导致泄漏。更广泛地说,最近在选择后推理方面的工作突出了执行统计分析的问题,例如聚类后的差异表达,即使聚类是在独立的数据集上定义的。

在交叉验证之外进行特征选择会产生不切实际的高模型准确性。演示了不恰当地使用监督特征选择方法(supervised feature selection approaches)对完全由随机值组成的合成数据集和标签向量产生的后果。
a|由于设计的数据没有真实的信号,一个经过特征选择的随机森林分类器在随机概率之上的表现并不显著(灰)。然而,当在交叉验证所有折叠之前错误地进行特征选择时,模型会随着特征数量的增加而表现出显著的增加和随后的性能下降(红)。这种有问题的行为发生在任何机器学习(ML)模型中。
b|使用监督特征选择对200个特征进行主成分分析(PCA)投影,并用类别标签着色(蓝色=阴性,红色=阳性)。尽管这两类之间缺乏真正的差异,但特征选择过程破坏了随后的无监督PCA方法。在ML分析的范围之外,预处理和可视化整个数据集不一定是有问题的。但如果随后选择适合ML模型,则只需要使用训练集重新进行预处理。
大多数ML工具包通过只从训练集中学习参数,然后独立地将转换应用于训练集和测试集,从而实现无泄漏地应用有监督和无监督转换。例如,标准化(standardization)就是用一个变量减去平均值,然后除以它的标准差。均值和标准偏差(standard deviation)是可以从训练集中学习并应用到训练集中的参数,这些参数可以重新用于变换测试集。这与在划分为训练集和测试集之前从整个数据集学习均值和标准偏差相反。对用户来说幸运的是,这个过程已经包含在scikit-learn的pipeline和transformer 的API中,以及R包caret的train()函数的preProcess参数中。
陷阱5: unbalanced classes(不平衡类)

不平衡的数据(unbalanced data)会给模型的训练和评估带来困难。如果训练集和测试集是平衡的,而预测集是不平衡的,那么测试集的性能将不能反映预测集的性能(左图)。无论使用平衡训练集还是非平衡训练集,测试集的不平衡性应该是预测集不平衡性的反映(右)。理想情况下,还应该使用能够处理不平衡的性能度量(performance measure)。
如果示例在结果值之间均匀分布,则监督学习任务是平衡的,否则是不平衡的。很少有真正的数据集是完美平衡的,基因组学中的一些问题表现出极端的不平衡。例如,当将ML应用于数百万个基因组窗口(genomic windows)以预测给定窗口是否包含增强子时,具有验证示例(阳性)的窗口可能占总数的约1%。在预测患者疾病风险方面,Khalilia et al.报告的疾病患病率从0.01%到29%不等。一项预测有害非编码变异(deleterious non coding variants)的研究对孟德尔病(Mendelian disease)使用了400个阳性和1400万个阴性,对复杂疾病使用了2000个阳性和140万个阴性。在这些情况下,模型面临着对多数分类学习过度而对少数分类学习不足的风险。当少数群体是主要关注的对象,并且假阴性(false negatives)可能代价高昂时,这尤其成问题,例如在通过医学扫描检测疾病或预测药物组合的副作用时。重要的是,不平衡的数据也会影响回归任务(regression tasks)的性能,在回归任务中,标签是连续的值而不是离散的类。
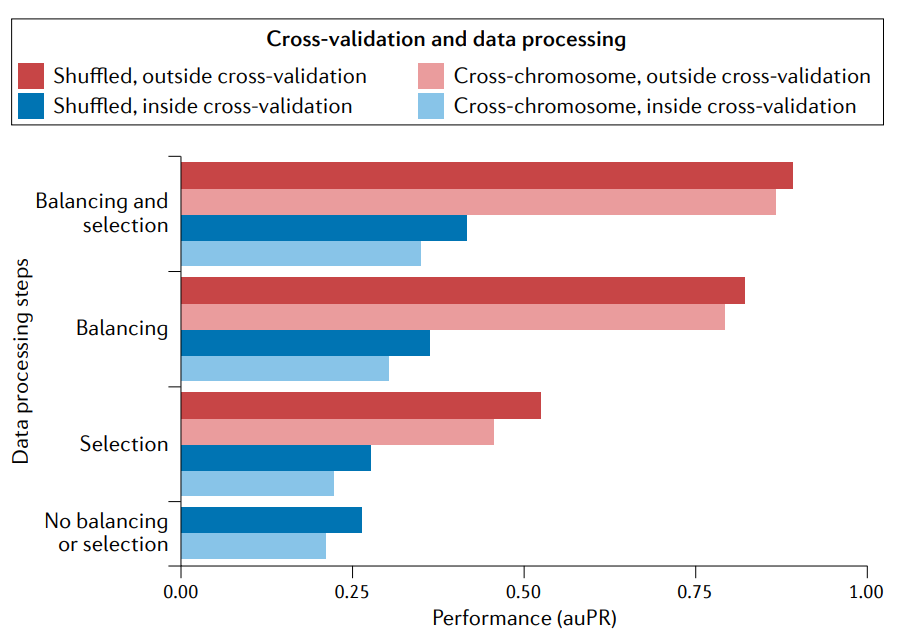
在交叉验证外应用平衡类可以提高性能。预测增强子基因组-宽(enhancers genome- wide)是一个很不平衡的分类问题例子,因为大多数基因组窗口不包含增强子。让阳性(有增强子)和阴性(无增强子)分类的规模相等(‘balancing‘)有助于交叉验证的训练。但是平衡应该在交叉验证(蓝色)中执行,以便用于评估性能的测试集反映出基因组中的实际不平衡。如果做不到这一点,性能就会膨胀(inflate)(红色)。类似地,当在交叉验证之外执行选择特性子集的预处理(‘selection’)时,由于泄漏会使性能膨胀,而在交叉验证之外执行平衡和选择会导致最膨胀的性能。
通过一系列策略来解决分类不平衡问题。在建模过程中,我们可以把先验概率分布(prior probability distributions)放到类上。更常见的是,研究人员使用再平衡方法(rebalancing approaches)来提高少数类别的性能。这里有三种基本策略:对少数类进行过采样(oversampling),对多数类进行欠采样(undersampling ),以及对例子进行加权(weighting)。每一种方法都强调了少数类模型的重要性。过采样要么重复现有数据(替换抽样),要么利用现有例子的插入(interpolation)来合成少数类的“可信的”(‘plausible’)例子。加权例子涉及使用类比例的逆(inverse),以便ML模型更强调少数类的误差。每一种方法都有不同的权衡(trade-offs):过采样保留了所有数据,但增加了计算时间;欠采样减少了计算时间,但丢弃了部分数据;样本加权保留了所有数据,但需要确定最优权重(optimal weights)。scikit -learn的imbalanced-learn包,以及caret中train的weights参数和trainControl的sampling参数,为Python和R的用户提供处理类不平衡的现成方法。
这三种方法都倾向于降低多数类的准确性,同时提高少数类的准确性,这通常是一种理想的权衡。然而,有两个重要的注意事项要记住。首先,平衡应该只在训练集折叠(training fold)内进行,以便根据预测集中预期的类别分布来评估拟合模型。其次,再平衡类别将导致估计器成为未校准(uncalibrated)的,即预测的概率分布将更接近于平衡训练集而不是不平衡测试集。当示例的排名比预测概率本身更重要时,这就不一定是一个问题,而且估计器可以使用后处理步骤(post processing step) 重新校准(recalibrated),但应该记住这个问题。
Conclusions
这篇综述主要说明了ML陷阱对模型性能的直接影响。然而,ML通常用于获得生物学观点(biological insights),而不是预测本身。在这些情况下,通常会解释一个训练好的模型,在确认它表现良好之后,提取它所学到的关系。不幸的是,如果一个或多个陷阱影响了分析,表现出良好性能的模型可能学习了无意义的关系。我们对检查陷阱的建议甚至对模型解释也很重要。然而,在某些情况下,过度拟合或有偏见的模型可以产生准确的生物学结论;不足于一般化的预测,但足够于归因,例如,由于混杂或依赖,模型可能会高估特征和结果之间的关联强度。当这是一种真正的生物学关系时,就会出现效应大小错误,但这种关联并不是错误的发现。
往期
