
https://doi.org/10.1038/s41592-022-01431-4
CAMI2宏基因组分析方法评估
在过去的20年里,宏基因组学的发展极大地增加了我们对人体和环境微生物组的了解,并促进了相关数据分析技术的发展。现如今,分析宏基因组数据的方法层出不穷,这就需要我们对这些方法进行公正和全面的评估,以方便我们为自己的数据选择和设计最佳的分析流程,得到最接近真实的研究结论。CAMI (Critical Assessment of Metagenome Interpretation) 就是为了满足以上需求而发起的大型合作研究项目。目前CAMI挑战项目的第二轮也就是CAMI 2已经结束,上百位领域内的科学家参与了评估分析来自不同环境(海洋,根系,多菌株混合,临床)的长读长和短读长复杂宏基因组数据集的方法,包括几十款宏基因组组装、分箱、序列分类、物种丰度预测、病原微生物鉴别软件。这些宏基因组数据集基于约1700个新的和已知的微生物基因组以及600个新的质粒和病毒创建。CAMI 2共分析了来自76个软件的5002个分析结果。
与第一次挑战中评估的软件相比,组装软件的性能最多提高了30%,总的来讲基于短读长的组装软件中HipMer, GATB的表型比较优秀(图1)。然而,在存在多个密切相关的菌株的情况下,组装的连续性、基因组完整度和菌株召回率都有所下降。这表明大多数组装软件,有时是故意的,没有解决菌株水平的组装,导致组装更零散,菌株特异性更差。此外,基因组覆盖率、参数设置和数据预处理影响了组装质量,而不同版本的软件性能相似。大多数提交的宏基因组组装只使用了短读长,而长读长和混合组装的总体质量并不高。但是,混合组装对于难以组装的区域,如16S rRNA基因,比大多数短读长组装的更加完整。混合组装软件也较少受到样本中密切相关菌株的影响,这表明长读长有助于区分菌株。
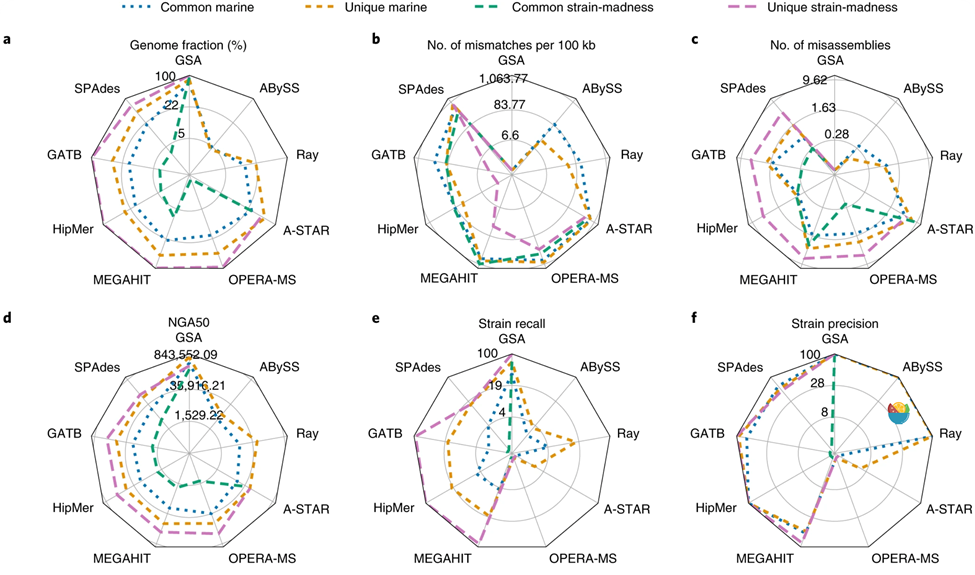
图1:宏基因组组装软件性能。a, 基因组完整度。b, 每100 kb的错配。c, 错误组装。d, NGA50。e, 菌株召回率。f, 菌株精度。线条表示所分析的基因组的不同子集,GSA (gold standard assembly) 的值表示了各个指标的上限。蓝色表示独特的基因组(与之最接近的基因组的ANI<95%),绿色表示常见的基因组(与之最接近的基因组的ANI ≥95%)。Strain recall = assembled high quality genomes / total genomes, Strain precision = assembled high quality genomes / assembled genomes.
与大多数单一分箱软件相比,集合多个分箱软件的方法(如MetaBinner, UltraBinner, MetaWRAP)在各种指标上有巨大的提升。单一分箱软件CONCOCT的表现也相当不错。总的来说,基因组分箱软件在不同的指标和数据集类型中表现出不同的性能,菌株的高多样性和较低的组装质量带来了巨大挑战,大大降低了性能。在植物相关微生物组数据集中,植物宿主和55个真菌基因组具有足够的覆盖率,因此获得了高质量的分箱结果。
序列分类软件中,MEGAN和Kraken的综合表现最佳。不少丰度预测软件如mOTUs与MetaPhlAn在第一轮CAMI挑战中就表现优异,在本次评估中他们同样表现优异(图2),属水平以上的分类性能很好,而在物种水平上这些软件性能大幅下降,同时对古细菌和病毒的表现也不理想。在临床病原体的挑战中,有几份提交的报告准确的给出了致病病原体。然而,这些结果都是由人工整理得到,没有一个是可重复的,这表明这些方法仍然需要大幅改进。尽管临床宏基因组学在病原体诊断和特征分析方面有很大的潜力,但各种各样的挑战仍然阻碍了其在常规诊断中的应用。
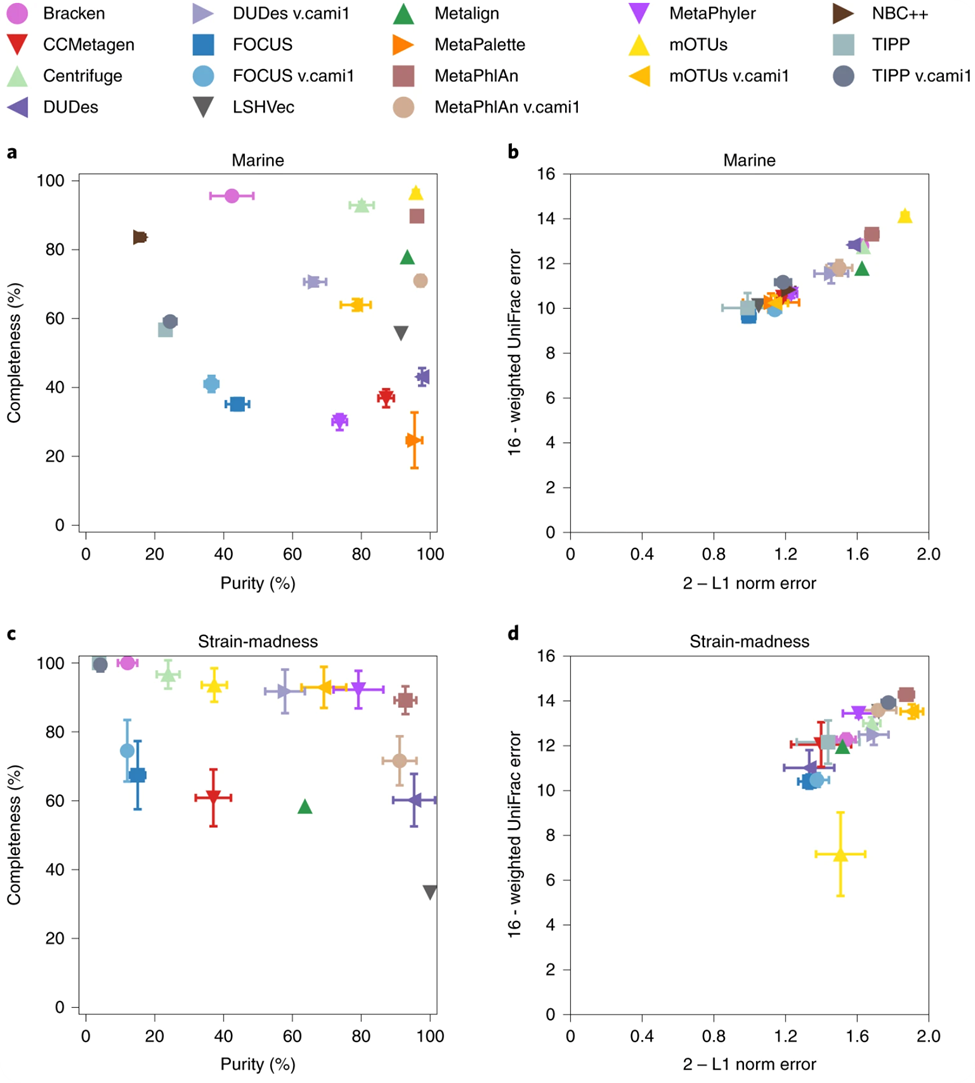
图2:属水平的海洋和多菌株混合数据集的分析结果a,b, 海洋数据集。c,d, 菌株混合数据集。
在第二次挑战评估中,CAMI展示和剖析了常见的宏基因组学分析软件的主要进展以及当前的挑战。随着方法和数据生成方面的继续发展,不断重新评估这些问题将显得非常重要。我们鼓励每个对微生物组研究中的基准测试、方法评估感兴趣的研究者加入CAMI挑战中以帮助微生物组学研究人员针对自己的数据和科学问题设计最佳分析流程。
Fernando Meyer, Adrian Fritz, Zhi-Luo Deng, David Koslicki, Till Robin Lesker, Alexey Gurevich, Gary Robertson, et al. 2022. Critical Assessment of Metagenome Interpretation: the second round of challenges. Nature Methods 19: 429-440. https://doi.org/10.1038/s41592-022-01431-4
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
点击阅读原文,跳转最新文章目录阅读