中国的肠道微生物群及其与主食类型、民族和城市化的关系
Chinese gut microbiota and its associations with staple food type, ethnicity, and urbanization
研究介绍
肠道微生物群对人类健康和疾病影响重大。目前我国仍然缺乏全国性的中国肠道微生物群基线研究。肠道微生物群的失衡(即生态失调)与许多疾病有关,然而,将微生物群研究转化为临床实践仍然受到多重挑战的限制。我们对来自8 个民族、居住在28 个省的63个县/市的2678 名健康中国人的粪便样本进行了16S rRNA 基因测序。我们确定了四种肠型,其中三种富含普氏菌、拟杆菌和埃希氏菌,而第四个没有显性通过评估肠道微生物群与属于6个类别的20个变量之间的关联,地理、人口统计学、饮食、城市化、生活方式和采样月份,我们发现地理解释了最大的微生物群变异,并阐明了与主食关联的不同模式食物类型、种族和城市/农村居住地。
材料与方法
- 采样:采集2678名来自中国不同区域的健康参与者的粪便样本

2)测序:16S rRNA测序,引物314F_806R
3)分析:
① 通过使用SEPP 算法将 ZOTU 代表序列插入到 99% Greengenes 13_8 参考树与 QIIME2 v2018.10 中构建系统发育树。
② 使用PICRUSt2 基于ZOTU 预测功能潜力,生成EC数和MetaCyc 通路丰度。
③ QIIME2 估计ZOTU、Faith’s PD、香农指数、Bray-Curtis 距离和未加权的 UniFrac 距离 phyloseq R v1.32.0 包估JSD
④ vegan R package v2.5–6的envfit函数评估由环境变量解释的微生物群变异,并使用PERMANOVA和vegan的adonis函数评估微生物群落的差异聚类。
⑤ 通过 Mantel 统计基于 Spearman 等级相关性估计地理距离和微生物 JSD 之间的相关性
⑥ 样本使用 cluster R 包 v2.1.0 的 pam 函数进行聚类
⑦ 使用 ade4 R 包 v1.7-15 在 PCoA 上可视化聚类
⑧ 使用 randomForest R 包 v4.6-14 进行十次五倍交叉验证的随机森林分析识别每个肠型的驱动属
⑨ 使用 scikit-learn Python 包 v0.23.1 应用具有十次五重交叉验证的随机森林模型来识别区分八个种族的属。为了平均每个族群中的个体数量,汉族人被随机下采样到完整数据集的 6% 1000 次。因此,将基尼系数、AUC、灵敏度、特异性和精确度的平均下降计算为 1000 个随机森林模型的平均值。
⑩ 为了检查微生物群共现网络,使用 SparCC 对属(数据精简为 10,000 个读数)进行相关分析,并使用 Cytoscape v3.5.1 59 进行可视化。
⑪ 除非另有说明,否则使用 DESeq2 R 包 v1.29.14 检测差异属,调整年龄和性别。除非另有说明,否则将平均相对丰度 > 0.1% 且在至少一组样品中存在 > 50% 的属用于上述分析。
结果
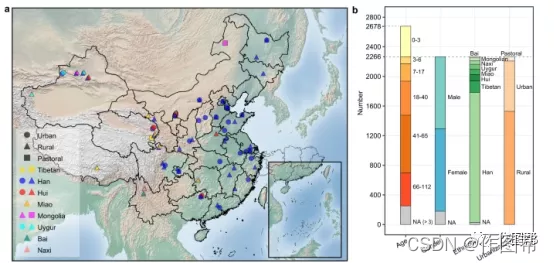
Fig. 1 研究对象概览. a)采样点、民族和城市化状况的地理分布。采样点是使用 Matplotlib 底图工具包绘制的。b)与年龄、性别、种族和城市化状况相关的队列构成。
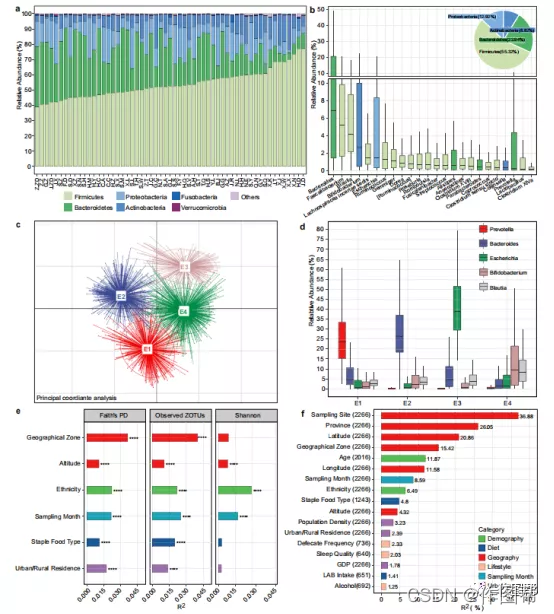
Fig. 2 中国人群的肠道菌群组成及相关协变量. a)每个采样点前六门的相对丰度。b)24个核心属的相对丰度,出现在 >90% 的个体中,平均相对丰度 >0.1%。饼图显示了该队列的门级微生物组成。LAB:乳酸菌。c)PCoA 图显示四种肠型。d)肠型代表性属的相对丰度。e)与微生物群α多样性相关的协变量。显示了具有 至少一个 alpha 指数的调整后的R 2 > 0.01 和 **** p < 0.0001(简单线性回归)的协变量。f)通过 JSD 距离估计的与微生物群 beta多样性相关的协变量。使用 envfit (vegan) 计算效果大小,并显示了p .adj < 0.05 的协变量。样本数量在每个协变量后面的括号中表示。在箱线图中,中心线代表中位数,箱线界限代表上下四分位数,胡须代表 1.5×四分位距。

Fig. 3 食用不同主食的人群之间的微生物群 alpha 多样性、组成和功能潜力的差异。a)观察到的 ZOTU。每个点代表一个采样点;点的颜色表示每个站点的中值;点的直径与每个站点中的样本数成正比,并且对于具有 >15 个样本的站点是固定的。b)三组之间的差异属。与属p .adj <0.05从DESeq2分析均显示。以红色或蓝色突出显示的属表示与小麦或大米摄入量相关的丰度较高,这是用两个 DESeq2 模型检测到的。R大米,W小麦。c)水稻和小麦组之间的差异糖苷酶。显示了来自 Mann-Whitney 检验的log2FC > 0.5 和p .adj < 0.05 的糖苷酶。d)水稻和小麦组之间的差异途径。通路按功能类别分组,小麦组中丰度较高的通路放置在图的上部。与L-甲硫氨酸和S-腺苷-L-甲硫氨酸生物合成相关的途径以红色突出显示。显示了来自 Mann-Whitney 测试的log2FC > 0.1 和p .adj < 1e-10 的路径。在箱线图中,中心线代表中位数,箱线界限代表上下四分位数,胡须代表 1.5×四分位距。大米:n = 417,小麦:n = 549,大米和小麦:n = 277。
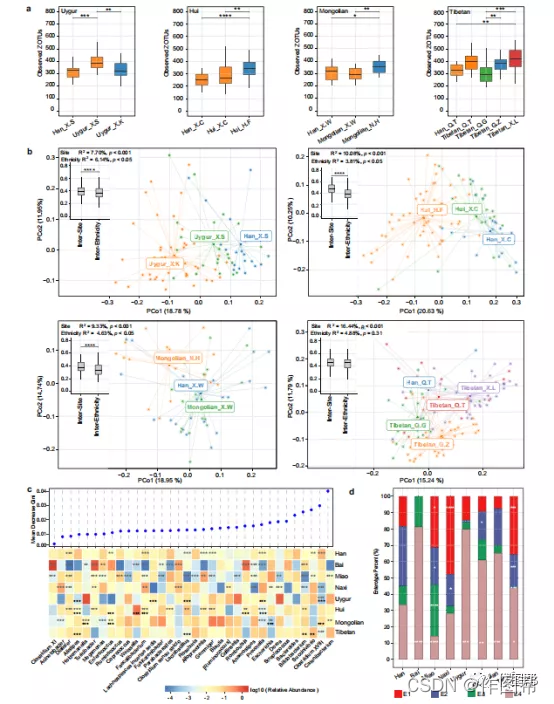
Fig. 4 不同种族的肠道菌群特征。a)每个采样点观察到的不同种族的 ZOTU。x轴的标签表示种族群体,然后是采样地点。* p .adj < 0.05,** p .adj < 0.01,*** p .adj < 0.001,Mann-Whitney 检验。b)基于 JSD 的 PCoA 图。插入的箱线图显示了属于同一种族的样本的站点间距离,以及来自相同采样站点的样本的种族间距离;**** p < 0.0001,Mann-Whitney 检验。还显示了来自 PERMANOVA 测试的相应R2和p值。c)族群间优势属丰度的差异。属按基尼系数的平均下降排序,这些模型来自对种族进行分类的随机森林模型。显示了平均相对丰度 >1% 和在至少一个族群中存在于 >50% 的样本中的属。星号代表来自 DEseq2 模型的p .adj 值;灰色表示一个民族与其他族群的比较(并根据采样点进行了调整),黑色表示每个少数民族(回族、蒙古族、藏族和维吾尔族)与其随附的汉族样本之间的比较相同的采样点;** p .adj < 0.01,*** p .adj < 0.001。d)每个种族的肠型百分比。p <0.05,* p <0.01,*** p <0.001,Fisher精确检验比较种族少数GROU ps到汉。在箱线图中,中心线代表中位数,箱线界限代表上下四分位数,胡须代表 1.5×四分位距。c – d汉族:n = 1755,白族:n = 16,苗族:n = 70,纳西族:n = 46,维吾尔族:n = 69,回族:n = 87,蒙古族:n = 40,藏族:n = 154。

Fig 5. 城乡人口微生物群多样性、组成和网络的差异。a)Faith’s PD。每个点代表一个采样点;点的颜色表示每个站点的中值;点的直径与每个站点的样本数量成正比,对于超过 15 个样本的站点是固定的。b)组间和组内 JSD。每个采样点计算组内距离。**** p < 0.0001,Mann-Whitney 检验。箱线图的中心线代表中位数,箱线界限代表上下四分位数,胡须代表 1.5×四分位距。c)使用 DESeq2 模型检测差异属。与属p .adj 0.05所示<。d)属的共现网络。SparCC 与r 的相关性 显示 > 0.35 和p < 0.01。每个节点重新p不满一个属; 节点的大小与相应属的中位相对丰度成正比;绿色、红色和蓝色分别代表共享属、农村特定属和城市特定属。实线和虚线分别代表正相关和负相关,边缘的粗细与r值成正比。红色和蓝色星号表示c 中显示的差异属,*** p .adj < 0.001。城市:n = 637,农村:n = 1530。
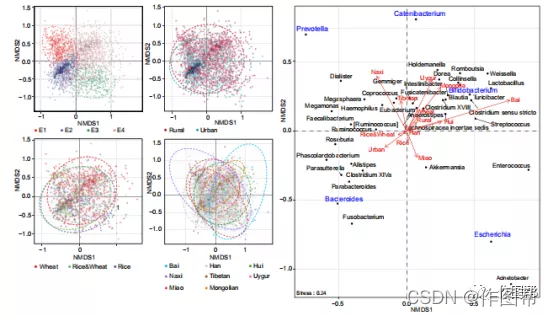
Fig. 6 非度量多维标度图显示了中国肠道微生物群按肠型、城市/农村居住地、主食类型和种族的聚类。排序是根据前 40 个属用 Bray-Curtis 距离进行的。使用 envfit (vegan) 将协变量从后投影到图上。椭圆代表 95% 的置信度。
END
(此推文仅供交流学习使用,侵权必删)