期刊名称:natrue communications
影响因子:12.298
日期:2021.5.24

研究介绍
结直肠癌(CRC) 是最常见的癌症之一,总体死亡率很高。肠道微生物组是一种基于粪便的非侵入性生物标志物,用于代谢疾病和癌症。许多研究报告称,肠道微生物组是 CRC 发生和发展的重要病因因素,并且已经确定了CRC一些粪便微生物标志物。然而,尚不清楚这些生物标志物是否可以准确检测腺瘤、早期CRC。目前对微生物组与结直肠腺瘤之间关联的了解是有限的,在多个人群中用于早期腺瘤诊断的可复制标志物仍然难以捉摸。因此需要探索和鉴定能够更准确有效地诊断结直肠腺瘤的基于粪便的微生物标志物。
材料与方法
2.1 取样
从复旦大学上海肿瘤中心的腺瘤患者、结直肠癌患者和健康对照者中收集粪便样本(招募患者进行初步诊断,并且在收集粪便样本之前从未接受过任何治疗。患有遗传性 CRC 综合征的患者和既往有CRC病史的患者被排除在研究之外)
将招募的受试者分为三组:
(1)健康受试者,即对照组:结肠镜检查肿瘤、腺瘤或其他疾病阴性的个体;
(2)腺瘤患者:结直肠腺瘤患者;
(3)CRC患者:初诊CRC患者。
最初招募总共94名受试者。除了相似的性别、年龄和 BMI 外,还基于纳入标准,招募了 43 名样本:30名CRC 患者、6名腺瘤患者和7名对照组。
2.2 公共数据搜集
从 PubMed.gov上已发表的研究中收集CRC患者、腺瘤患者和健康对照的16S rRNA 测序数据。
2.3 数据预处理
QIIME2 (V.2018.11) 分析16S rRNA 测序数据。
2.4 混杂分析:
使用ANOVA 分析来量化潜在混杂因素和疾病状态的影响。
2.5 差异丰度ASV 的Meta 分析:
使用R (V.3.5.2)“coin”包中实施的两侧Wilcoxon 秩和检验对在单个ASV上差异丰度的显著性进行测试(P值< 0.05 被认为在所有差异分析中都具有显着性)。
2.6 模型构建和特征提取
用scikit-learn (V.0.19.2)包构建RF模型,并使用分层10倍交叉验证将腺瘤与癌症或对照区分开来。用于模型构建的特征包括患者元数据以及差异ASV和alpha多样性指数。
2.7 模型评估
进行研究间转移验证和 LODO 验证,评估基于微生物的腺瘤分类器在不同背景下的通用性,例如地理变异和微生物数据生成和处理在多个患者人群中的技术差异。
2.8 共现和聚类分析
使用SparCC算法对差异 ASV 的相对丰度进行了网络分析。P 的相关系数值< 0.05(定义为显著)和大于0.1(对照与腺瘤)或大于0.3(腺瘤与癌症)的值被选择用于Cytoscape (V.3.8.0) 中的进行更层次的可视化。模块化结构和高度互连的节点组使用标准参数的MCODE 应用程序进行分析。
2.9 FIT对大肠腺瘤的诊断能力
从已发表的研究中收集了公开可用的 FIT 样本(包括172 名对照个体和198 名腺瘤患者),评估传统非侵入性测试FIT 的诊断能力。
2.10独立研究和非CRC疾病的额外验证
使用与发现RF模型相同的超参数重建了RF模型,使用所有具有相同分类分配(在属级别)的ASV 以及患者元数据(由于缺乏患者元数据,仅将ASV 用于验证队列)作为输入特征。这些特征的性能在五个非CRC疾病(CD,UC,IBS,NAFLD,T2D)的表现,对于每种疾病,构建RF模型来区分非crc疾病和对照组。
2.11功能概况分析
从 PICRUSt2 (V.2.0.3-b)的16S rRNA 序列推断出肠道微生物组的功能。
2.12 qRT-PCR 验证
对7个健康对照、6个腺瘤和30个CRC 样本进行了三次qRT-PCR 分析,量化来自两个选定生物合成的基因的丰度和表达。
结果
3.1 荟萃分析中数据集的特征
总共收集了来自结直肠腺瘤患者的306个样本、来自CRC受试者的217个样本和来自健康对照的252个样本,调查了来自四项研究的16SrRNA测序数据,以评估随着CRC进展(从对照到腺瘤再到癌症)的肠道微生物组变化,并确定腺瘤特有的生物标志物。表1包含了本研究中包含的大规模腺瘤数据集的特征信息。
3.2 元分析中潜在混杂因素的识别及结直肠腺瘤肠道微生物组成的变化
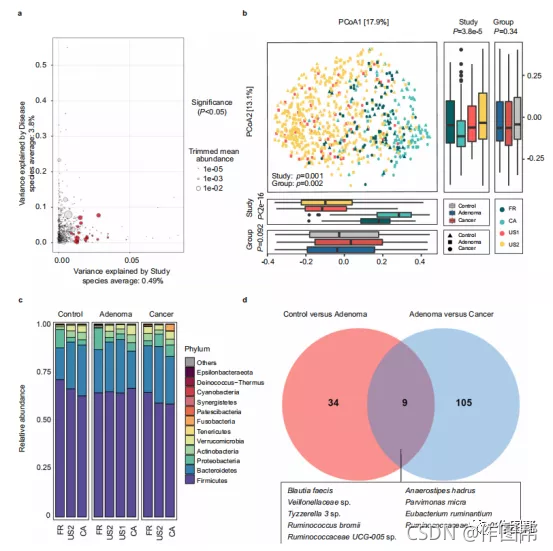 图 1:不同疾病状态下肠道微生物组成的变化。
图 1:不同疾病状态下肠道微生物组成的变化。
a由疾病状态(腺瘤与癌症)解释的方差与由个体ASV的研究效果解释的方差作图。显著差异的ASV 用红色表示,点大小与每个ASV的丰度成正比。P值来自Wilcoxon 秩和检验。源数据和精确的P值作为源数据文件提供。b 基于Bray-Curtis 距离对来自所有四项研究的样本(对照,n = 252;腺瘤,n = 306;癌症,n = 217)进行主坐标分析,这表明研究之间的粪便微生物群组成不同(P= 0.001 )和组(P= 0.002)。使用PERMANOVA 计算基于Bray-Curtis 距离的beta 多样性的P值。该组(对照组、腺瘤和癌症)用不同的形状表示。右上角和左下角的箱线图说明了投影到前两个主坐标上的样本,分别按研究和疾病状态细分。第一和第二主成分的P值采用双侧Kruskal-Wallis检验计算。所有箱形图代表分布的第25-75百分位;中位数显示在框中间的粗线;须延伸到IQR1.5倍以内的值,异常值用点表示。源数据作为源数据文件提供。c四项不同研究中健康对照、腺瘤和CRC中细菌门的相对比例。d维恩图显示了腺瘤和健康对照或CRC之间在物种水平上分配的差异ASV 的重叠。
3.3 结直肠腺瘤微生物分类模型
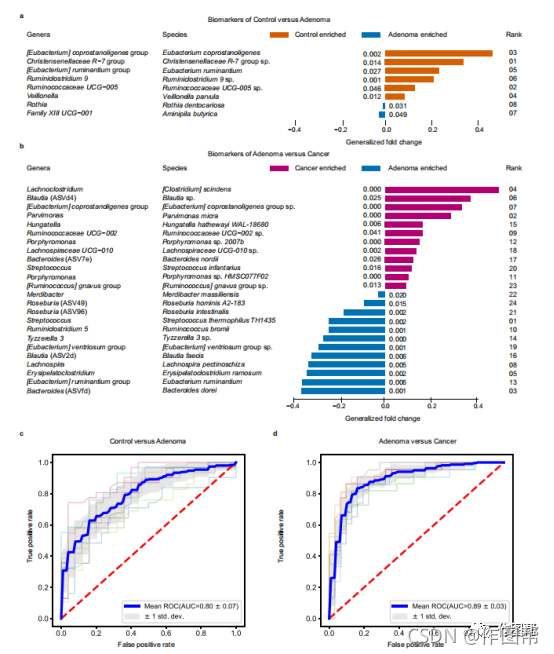 图 2:使用重要特征区分腺瘤与对照或癌症的性能。
图 2:使用重要特征区分腺瘤与对照或癌症的性能。
a,b,鉴定生物标志物以构建用于区分腺瘤与对照(a)和CRC(b)的RF模型。每个生物标志物代表一个ASV,属和种列显示了在属和种水平上的ASV的分类信息。。a和b中的rank表示RF模型中特征重要性的顺序;P值是使用双侧阻塞Wilcoxon秩和检验计算的,精确的P值显示在条形图旁边。广义倍数变化(参见差异丰度ASV的荟萃分析方法)由颜色梯度表示。源数据作为源数据文件提供。c , d优化模型的AUC由生物标志物和对照与腺瘤©和腺瘤与癌症(d)的患者元数据构建。分层10倍交叉验证的平均AUC和标准偏差显示在c和d 中。
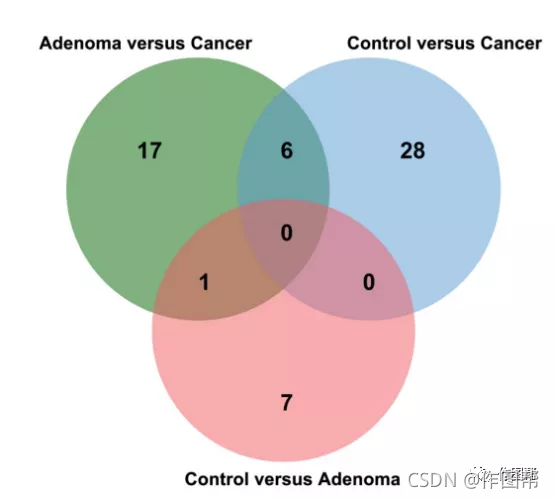
补充图 5|维恩图中三组生物标志物的重叠。
3.4 微生物群的共现和聚类分析
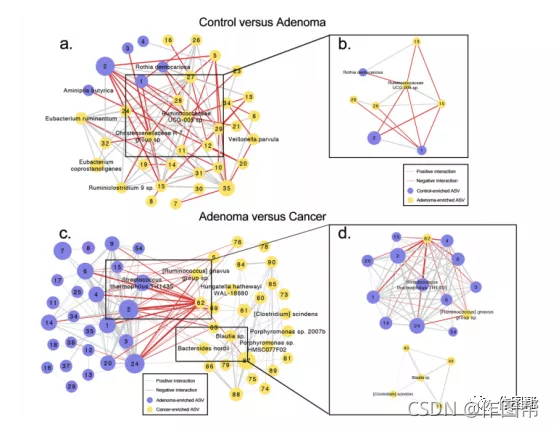 补充图 6|生物标志物的微生物相关网络。(a)腺瘤与对照组之间差异ASVs之间的相关网络(n=43差异ASVs)和©腺瘤与CRC之间差异ASVs的相关网络(n=117差异ASVs)。相关系数采用SparCC算法计算。模块(b)和(d)分别使用(a)和©中的MCODE应用程序构建。节点大小代表平均ASV丰度;生物标记物ASV被标注为物种;其他差异ASV用节点数表示;边缘表示相关性:边缘厚度表示大小,颜色表示相关性的符号(灰色、正,红色、负)
补充图 6|生物标志物的微生物相关网络。(a)腺瘤与对照组之间差异ASVs之间的相关网络(n=43差异ASVs)和©腺瘤与CRC之间差异ASVs的相关网络(n=117差异ASVs)。相关系数采用SparCC算法计算。模块(b)和(d)分别使用(a)和©中的MCODE应用程序构建。节点大小代表平均ASV丰度;生物标记物ASV被标注为物种;其他差异ASV用节点数表示;边缘表示相关性:边缘厚度表示大小,颜色表示相关性的符号(灰色、正,红色、负)
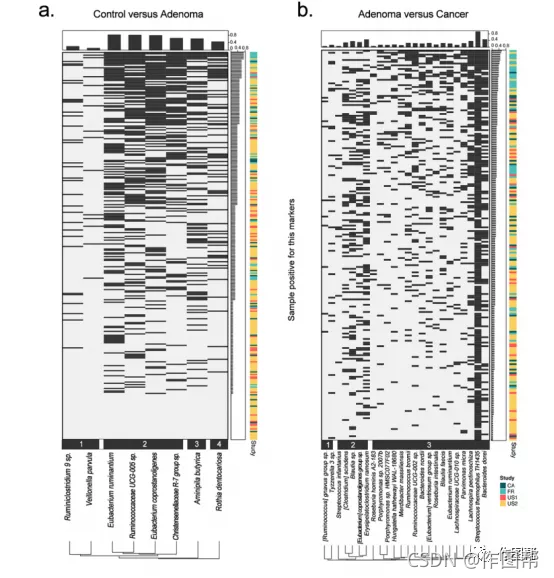 补充图 7|区分腺瘤与对照组或结直肠癌的生物标志物的共现分析。对于所有(a)腺瘤(n=306)或(b)CRC(n=217)的患者,热图显示了各自的样本是否对每个生物标志物都呈阳性。样本按阳性生物标志物的和排序,根据阳性样本的Jaccard指数将生物标志物分为4个聚类(a)或3个聚类(b)。
补充图 7|区分腺瘤与对照组或结直肠癌的生物标志物的共现分析。对于所有(a)腺瘤(n=306)或(b)CRC(n=217)的患者,热图显示了各自的样本是否对每个生物标志物都呈阳性。样本按阳性生物标志物的和排序,根据阳性样本的Jaccard指数将生物标志物分为4个聚类(a)或3个聚类(b)。
3.5 结直肠腺瘤分类器的验证
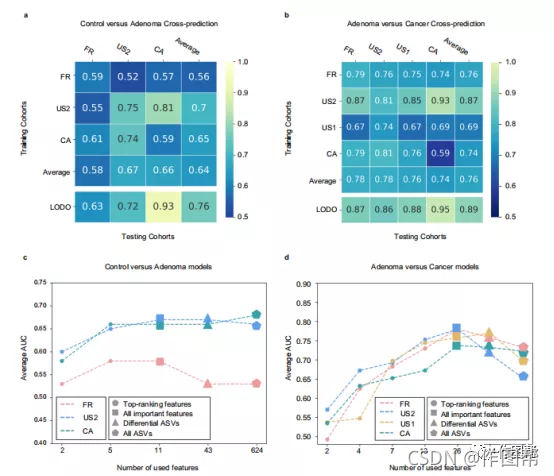
图 3:跨研究的重要特征的预测性能和用于检测腺瘤的最小特征的识别。
a,b,交叉预测矩阵详细描述了使用套袋k-最近邻分类器(a)和CRC使用RF模型(b)来区分腺瘤和对照的AUC。对角线上的值是指每个研究中的交叉验证结果。非对角线值是指交叉队列验证得到的AUC值,对分类器进行相应行的研究训练,并将其应用于相应列的研究。LODO值是指除了使用相应列的研究外的所有方法训练分类器并将其应用于相应列的研究所获得的性能(见“模型评估”部分)。使用RF模型鉴别腺瘤与对照组的研究到研究和LODO验证值可以在补充图中找到(补充图 9)。c,d具有不同特征集的对照与腺瘤©和腺瘤与癌症(d)的研究到研究的转移验证分类器的平均AUC。输入特征分别表示为不同的形状、排名靠前的特征、所有重要特征符号、差分asv和所有asv分别用圆、正方形、三角形和五边形表示。c和d中的x轴表示不同数量的特征。颜色代表不同的研究。
3.6 独立队列中结直肠腺瘤标志物的验证
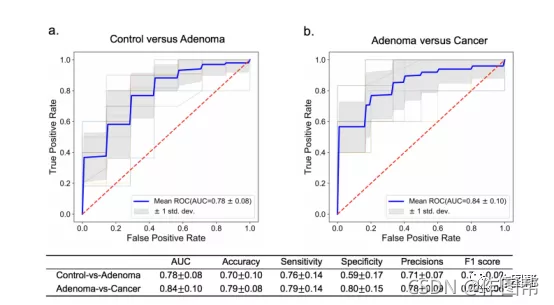
补充图 15|验证两个独立队列鉴别腺瘤与对照组(a)和CRC(b)的性能
3.7 结直肠腺瘤预测模型的特异性
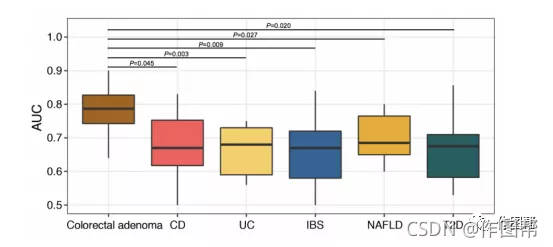 补充图 16|腺瘤预测模型的特异性。
补充图 16|腺瘤预测模型的特异性。
比较不同微生物组相关疾病模型的重要特征的性能:腺瘤(n=102)与对照(n=70)模型、乳糜泻(n=61)与对照(n=18)模型、UC(n=47)与对照(n=18)模型、IBS(n=84)与对照(n=44)模型、NAFLD(n=18)与对照(n=51)模型、T2D(n=48)与对照(n=214)模型。所有箱形图代表分布的第25-75百分位;中位数显示在盒子中间的粗线表示;须在1.5倍IQR内延伸到最极端的点。P值采用双侧Wilcoxon秩和检验计算。
3.8 结直肠腺瘤的微生物功能变化
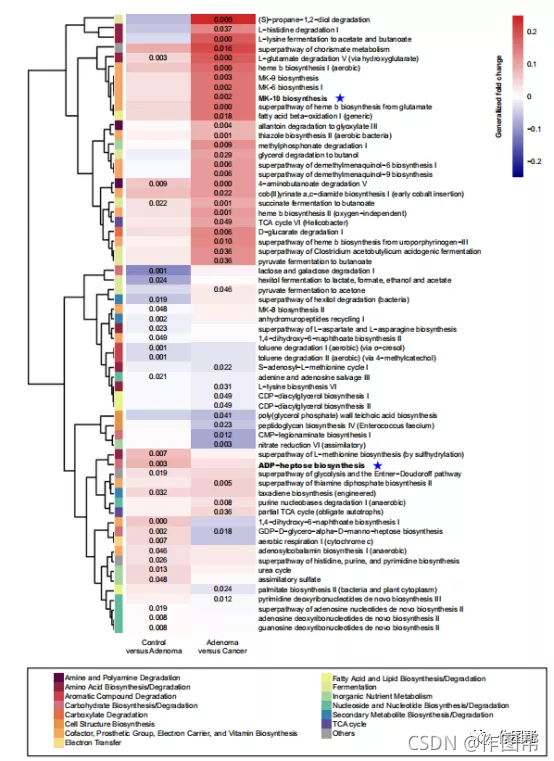
图 4:对照、腺瘤和癌症的功能改变。
比较了腺瘤与对照组或癌症之间功能通路的相对丰度。绘制了差异丰富的路径;P值采用双侧阻塞Wilcoxon秩和检验计算,热图中给出了准确的P值<0.05。广义褶皱变化(见“差异丰富asv的meta分析”部分)用颜色梯度表示。广义折叠变化>0:后者富集;广义折叠变化<0:前者富集。
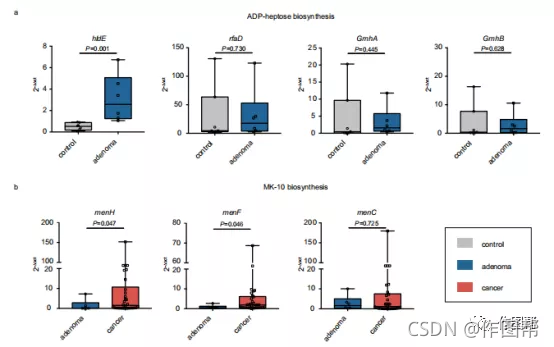
图 5:候选基因的相对丰度。绘制的值是adp-七糖和MK-10生物合成中细菌基因的qRT-PCR定量。比较对照组(n=7)和腺瘤(n=6)组的hldE、rfaD、GmhA和GmhB的丰度,同时比较腺瘤(n=6)和癌症(n=30)组的bmenH、menF和menC的丰度。所有的盒子从第25百分位延伸到第75百分位,须显示最小值和最大值。每个盒子中间的线条显示了中位数。P值采用双侧Wilcoxon秩和检验计算。
讨论
(1)粪便细菌可作为生物标志物用于CRC
本研究综合评估了CRC相关肠道微生物组的改变以及微生物标志物在癌前阶段腺瘤早期检测CRC的能力。长期以来,人们一直报道了粪便细菌可作为生物标志物用于CRC,如非侵入性诊断具核梭杆菌,大肠埃希氏菌,以及脆弱拟杆菌。然而,这些微生物标记的研究之间存在很大差异,表明多队列整合分析的必要性。两项开创性研究已经进行了跨队列分析,重点是根据 WMS 数据区分CRC 患者和对照组。相比之下,我们的研究旨在识别腺瘤特异性微生物标志物,因为早期筛查CRC 对患者至关重要。总体而言,本研究中的这些验证都表明基于粪便的微生物标记物可以作为有效的非侵入性临床指标用于结肠直肠腺瘤。
(2)结直肠腺瘤和癌症的微生物群落各不相同
在结直肠癌进展过程中,结直肠腺瘤和癌症的微生物群落各不相同。研究结果表明 CRC 相关生物标志物对检测结直肠腺瘤无效,并强调了腺瘤特异性特征的重要性。此外,腺瘤特异性标志物可能有助于早期筛查,从而降低CRC 的风险。无创FIT测试可作为肠道微生物群分析的补充工具,用于腺瘤的早期筛查。
(3)功能分析揭示的潜在机制有利于进一步解释CRC致癌作用
功能分析揭示了复杂的潜在机制,并将大大增强我们对CRC致癌作用的理解和解释(补充图 17)。特别是,我们发现与对照相比,腺瘤中ADP-庚糖和关键基因hldE的生物合成显着富集。ADP-庚糖已被鉴定为细菌相关致癌物和LPS 生物合成中的关键代谢中间体。它是激活 NF-κB 信号传导的有效触发器,已被证明可促进肿瘤发生并且可能对持续炎症至关重要.从对照到腺瘤再到结直肠癌的ADP-庚糖生物合成途径的丰度增加表明该途径的活性升高可能是导致结直肠癌发展过程中NF-κB 信号传导持续恶化的一个重要因素。值得注意的是,与对照相比,腺瘤中ADP-庚糖生物合成的途径丰度显着增加,而与腺瘤相比,CRC中没有显着富集。这可能表明ADP-庚糖在腺瘤中发挥了关键作用,并在CRC进展中保持了这种作用。此外,一系列维生素K2生物合成基因,如menH和menF腺瘤和癌症之间也有显着差异。
(4)经多个研究可确定用于区分腺瘤与健康对照和结直肠癌的微生物来源标记物。
为了弥补没有设计干预研究来证明该论文这一弱点,我们努力从研究设计的其他角度加强证据,并对已鉴定的腺瘤微生物生物标志物提供不同类型的验证,以便早期检测CRC。综上所述,通过广泛和统计上严格的验证,我们在多个研究中确定了用于区分腺瘤与健康对照和结直肠癌的微生物来源标记物。独立验证证实微生物来源的标志物在检测腺瘤方面表现出高度的准确性和特异性。这些微生物来源的标志物可能有助于结直肠腺瘤的非侵入性诊断,并可以靶向抑制结直肠癌的发生。此外,我们提出微生物组的改变介导的ADP-庚糖生物合成激活了腺瘤中的炎症,而紊乱的微生物组通过增加CRC 致癌过程中维生素K2的产生而起到补偿作用。
(此推文仅供交流学习使用,侵权必删)