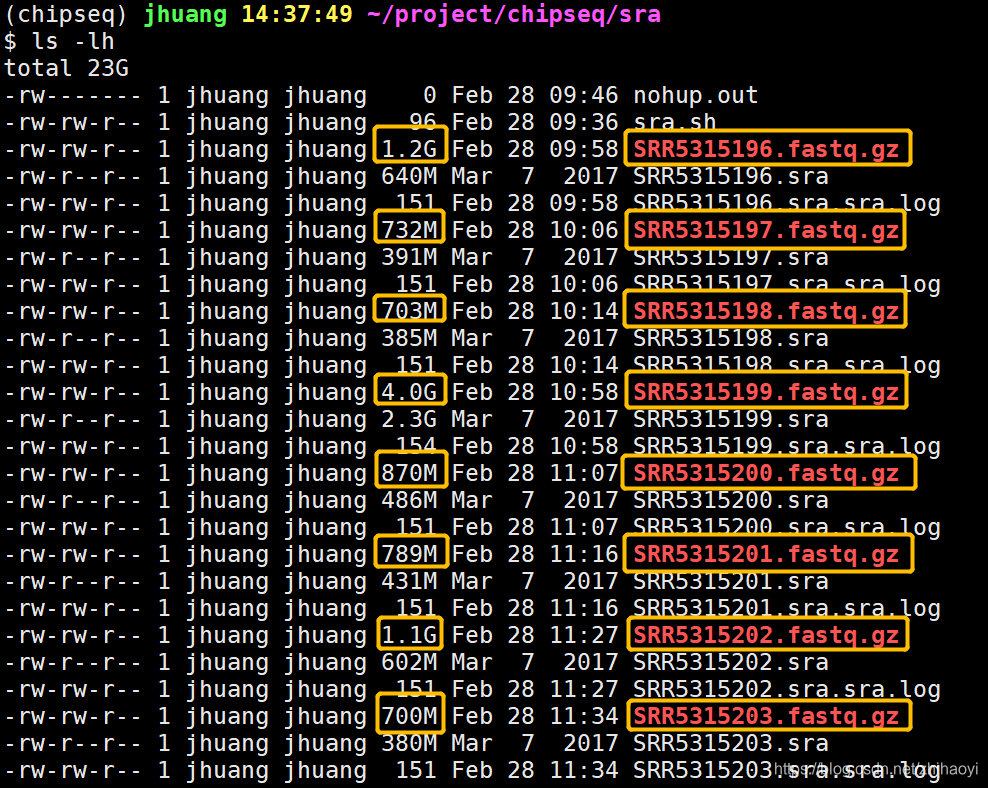
把sra数据移动到我们工作目录后,我们开始sra转faq。
正式运行代码之前,必须先拿一个样品测试下代码能否运行成功,这点很关键,因为这步就算成功运行也特别慢,要是代码再出错了就更浪费时间了。
拿第一个样品做测试
ls SRR5315196.sra |fastq-dump -gzip --split-3 -O ./ SRR5315196.sra
sra-tools 里的 fastq-dump工具可以将SRR文件转换为FASTQ格式,–split-3参数表示如果是双端测序就自动拆分,如果是单端不受影响。也就是说,–split-3参数可以将PE的sra文件解压后的fastq文件拆分成_1.fastq和_2.fastq,如果示例数据集是SE测序,不会进行拆分。–gzip转换fastq为压缩文件,节省空间。
单个测试成功,那我们就写循环进行批量转换格式。
cat >sra.sh #写脚本 ls SRR* |while read id; do (fastq-dump -gzip
–split-3 -O ./ {id}.sra.log 2>&1;done #多个循环 nohup bash sra.sh & #挂后台运行
这步一定一定记得挂后台运行(nohup cmd &)因为特别慢,不挂后台你一掉线就功亏一篑了。
我们解读下这个写入脚本里面的循环命令:
ls SRR* 是为了能够把当前文件夹下所有从NCBI上下载的SRR数据列出来
 fastq-dump -gzip --split-3 -O ./ ${id} 这条命令是告诉系统,我要转换成fastq格式并且要压缩的;而且表示如果是双端测序就自动拆分,如果是单端不受影响;-O ./
{id}.sra.log 2>&1 (0.标准输入;1.标准输出;2.标准错误)这个命令是说,重定向标准输出到当前文件下SRR5315197.sra.sra.log文件,且标准输出、标准错误到一个文件中(2>&1)
fastq-dump -gzip --split-3 -O ./ ${id} 这条命令是告诉系统,我要转换成fastq格式并且要压缩的;而且表示如果是双端测序就自动拆分,如果是单端不受影响;-O ./
{id}.sra.log 2>&1 (0.标准输入;1.标准输出;2.标准错误)这个命令是说,重定向标准输出到当前文件下SRR5315197.sra.sra.log文件,且标准输出、标准错误到一个文件中(2>&1)
 制作config文件
制作config文件
制作config文件的作用: 从NCBI上下载SRA数据,之后再转成 fastq.gz格式,在此过程中把原本的文件名(SRR号)改成在跑流程时可以区分各样本的文件名,生信分析中 文件命名很重要,生成fastq文件时,如果不进行更改操作就会直接生成 SRR****.fastq.gz ,我们不能这样,只有这些SRR号我们根本不知道这些样品是些什么,生信分析很重要的一点就是从文件名上我们就要知道这是个什么数据。
首先还去NCBI下载SRA数据的那个界面下载个 txt 文件
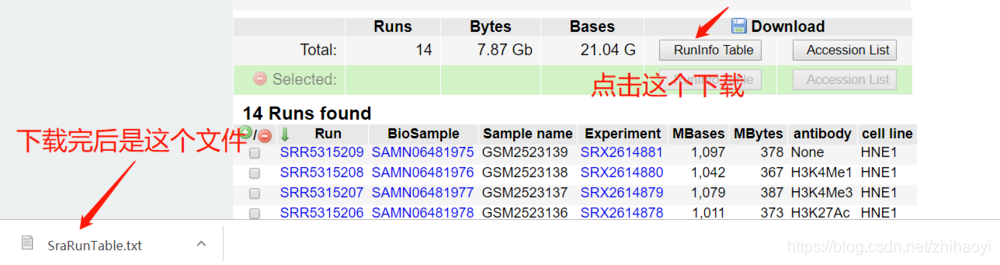 下载到自己的电脑上后,导入到服务器上
下载到自己的电脑上后,导入到服务器上
导入成功后,使用命令:
$ head -1 SraRunTable.txt | tr ‘\t’ ‘\n’|cat -n
 查看该文件的第一行(表头)把它转化成列,并加上行号。
查看该文件的第一行(表头)把它转化成列,并加上行号。
