生物医学大数据-蛋白质基因组学:质谱注释
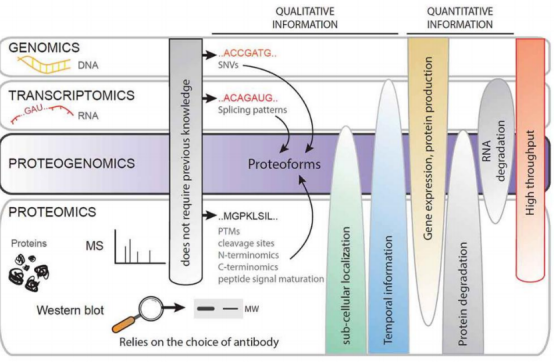
蛋白质组与其他组学的关系便是互为印证:蛋白质基因组学原本用于基因组注释,后面扩展到蛋白质与转录组或可变剪接之间关系,同时,蛋白质组依赖于基因组注释作为验证。许多研究未标明蛋白质基因组学,而是归属于对应的组学。
蛋白质基因组学现存问题:
基因组注释方法:1.Denovo。2.与转录组相应证。3.与基因组数据库同源比对。
基因组注释问题:
对于特定结构:
- 起始位点定位信号难以确定
- 确定promoter调控结合位点,错误率高
- 终止位点不被注释
- 可变剪接预测和确认上需要更多基因组的训练数据,使用denovo可以全部预测出来,但是用内含子和外显子仅能预测60%。
对于基因组的常规部分:
- 因为二代reads短所以难拼接
- 以前是鉴定已经存在的基因组,现在是未知物种的genome
- 同类数据不同处理有不同结果
- 人员水平不齐
对于蛋白质组大量质谱数据没被充分解析,可能的原因是
- 蛋白质多种修饰
- 搜索引擎差异
- 其它电荷没鉴定
- 数据库数据不足
没有充分解析造成许多没鉴定出来的数据,这些数据有可能是
- 噪音
- 实验污染,通过加入污染蛋白证实这部分是实验污染,当然鉴定出来之后也会舍弃这部分,对于该部分问题需要改进质控方法。
- 产生混合图谱(即多肽段在同一个图谱)或者融合肽段
- 在已知信息库中未收录的蛋白,即新蛋白
蛋白质基因组学的作用:
1.修正基因模型,即增加新注释,增加新肽段2.反过来由新肽段增加新基因
蛋白质基因组学需要提高的方法
蛋白质实验方面:
- 提高分离技术使蛋白质分的开
- 提高富集技术使得蛋白质量变大
- 高精度仪器
- 提高样本多样性,收集来源于不同时间空间的样本
数据库及数据处理方面:
常规部分存在的问题是大数据库存有大量噪音和相似,数据六位翻译结果搜索空间和denovo差不多。可以通过限制外显子大小,保留高分和正负库方法改进。质量评估时采用多段鉴定。假阳性高,动态范围宽,难鉴定可变剪接(可变剪接来源于denovo大数据库),重拼接假阳性高。
搜索速度方面:
质量过滤低质量、图谱聚类、去重或计算机并行的方法改良检索速度。
新方法:
加入RNAseq比对多重验证。
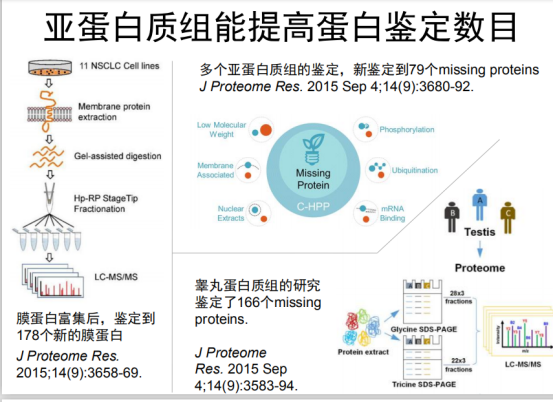
分成亚细胞组分多种方法鉴定。
体外转录翻译来验证

n-末端组学:使用对角线色谱查肽段末端

DNA和RNA数据辅助蛋白质鉴定

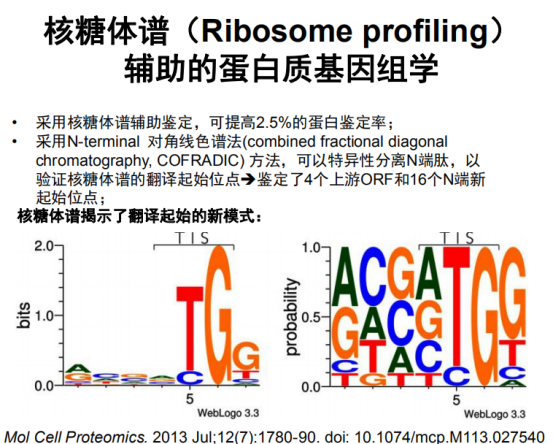
核糖体谱