一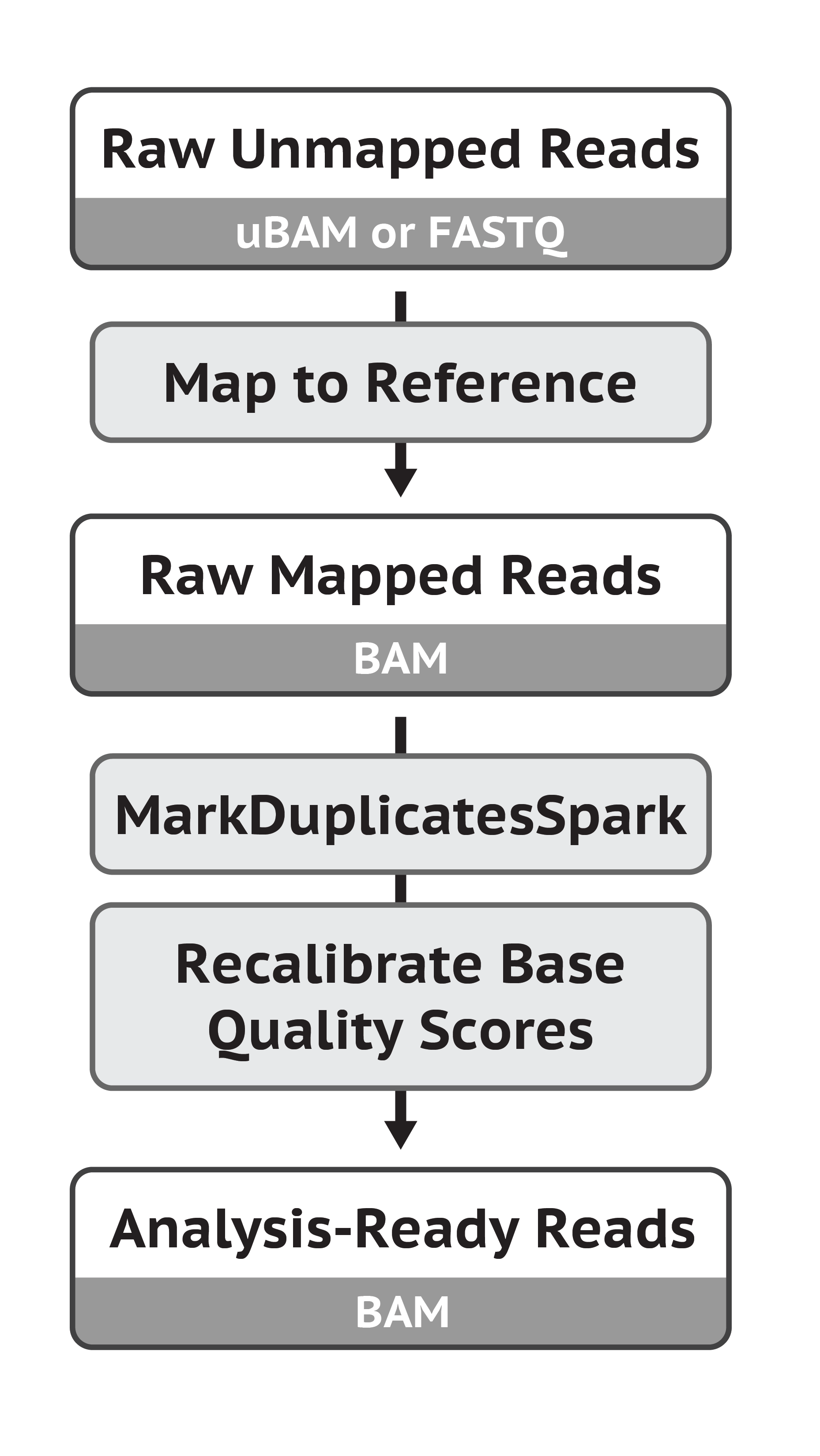
Pegasus是一个Workflow Management System。
利用它,设计一个DAG图作为一连串任务的流程,方便的进行并行、串行,并可从错误中恢复。
二、更多信息
https://pegasus.isi.edu/about/
三、GATK
Gatk Best Practice:https://software.broadinstitute.org/gatk/best-practices/workflow?id=11165
① 将Reads比对到参考基因组上:BWA
bwa mem ref.fa read1.fq read2.fq > aln-pe.sam
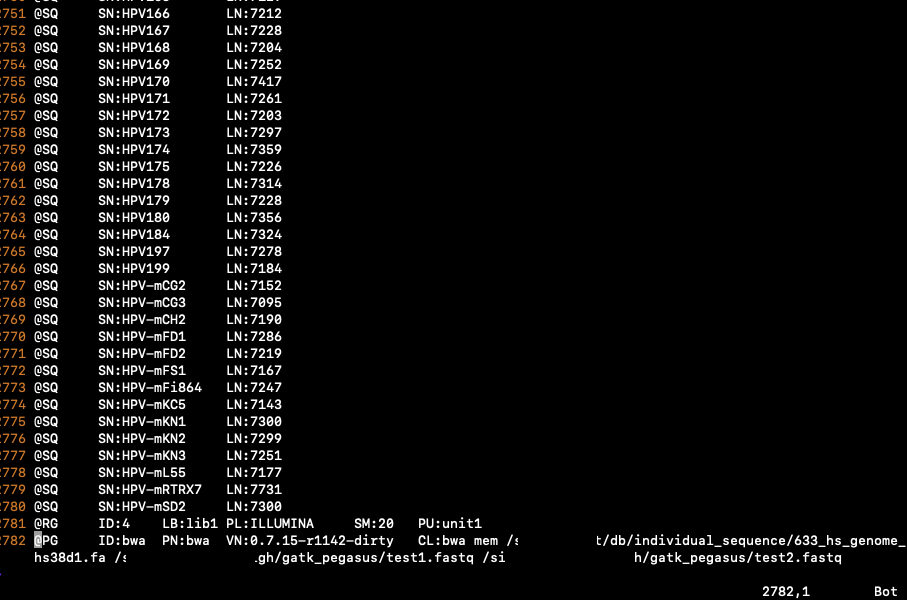

SN: Reference sequence name. The SN tags and all individual AN names in all @SQ lines must be distinct. The value of this field is used in the alignment records in RNAME and RNEXT fields. Regular expression: [:rname:∧*=][:rname:]*
LN: Reference sequence name. The SN tags and all individual AN names in all @SQ lines must be distinct. The value of this field is used in the alignment records in RNAME and RNEXT fields. Regular expression: [:rname:∧*=][:rname:]*
这步生成sam文件,然后要利用samtools将sam转为bam
SAM:Sequence Alignment Map
BAM:Binary Alignment Map
https://samtools.github.io/hts-specs/SAMv1.pdf
如果是将fastq文件分成多个来bwa的,现在要用picard将他们合并
java -jar picard.jar MergeSamFiles <argument...>
②Mark Duplicates:MarkDuplicatesSpark
- Reads are sorted into coorrdinate-order
- Mark the Duplicates, causing the marked pairs to be ignored by default during the variant discovery process.
它的意义参见 https://www.jianshu.com/p/cad6a359a368:SNP的发现


欢迎关注"生信修炼手册"!在数据预处理中,有一个很重要的步骤就是MarkDuplicates, 字面意思就是标记重复序列。重复序列是如何产生的,为什么要标记重复序列呢?首先来看重复序列产生的途径,有以下两种 PCR duplicates这个很好理解,PCR根据一份模板,扩增出多份拷贝,来源于同一模板的多份拷贝之间就是PCR重复序列 Optical duplicatesillumina测序仪的基本单位是flowcell,测序反应在flowcell上发生和进行,高密度的flowcell使得测序的通量显著提升,也带来了序列重复读取的问题。虽然比例非常低,但是也需要考虑进来。 GATK官方对PCR重复和系统重复进行了统计,可以看到,PCR重复的比例随着测序量的增加而增加,而Optical duplicates 重复序列的比例是一个随机分布,总是存在的,其比例相对稳定,在是在一定范围内波动,符合系统误差的特性。之所以要标记重复序列,是为了下游的SNP分析。SNP位点的识别,简单理解可以看做一个概率问题。比如下面两种情况: 情况A基因组上某位点碱基为A, 有100条reads 覆盖到该位点。 其中99条都为A, 1条为C; 情况B基因组上某位点碱基为T, 有100条reads 覆盖到该位点。 其中54条为T, 46条为C; 上述两种情况都检测到了两种碱基,是不是意味着检测到了两个SNP位点呢?当然不是,情况A中C碱基的比例为1%,很可能是测序错误,当然不能算是一个SNP位点;情况B只从reads分布看,可以认为是一个候选的SNP位点,当然还要分析其他的因素才能判断是否是一个snp位点。从这里也可以看出, reads 的计数对于SNP位点的检测特别的重要。但是这里的reads 指的是有效reads , 是实际在样本中存在的reads的数目。在计数时,重复序列只计数1次。MarkDuplicates的作用就是标记重复序列, 标记好之后,在下游分析时,程序会根据对应的 tag 自动识别重复序列。重复序列的判断方法有两种: 序列完全相同 比对到基因组的起始位置相同 序列完全相同时,认为是重复序列当然没什么大问题。虽然会有同源性,重复序列等因素的影响,但是概率非常之小,基本上可以忽略不计;比对位置相同也认为是重复序列,是因为在测序过程中,会存在测序错误,本身完全一样的序列, 可能测序得到的的reads并不完全相同(可能有几个碱基不同),而且在去除低质量的过程中,也会有所差异(末端切除的低质量碱基数不同), 所以最终根据比对基因组的结果进行判断。如果序列比对到基因组上的起始位置是相同的,就认为是重复序列。GATK4 标记重复序列的命令如下:soft/gatk-4.0.4.0/gatk MarkDuplicates -I input.bam -M metrc.csv -O marked.bam在输出的bam文件中,借助第二列的flag 来标记重复序列,flag的值是多种情况的叠加,其中1024代表重复序列 samtools flags 1024 0x4001024DUP 在生出的bam文件中,通过flag的值可以知道该序列是否为重复序列。通过flag已经可以知道哪些是重复序列了,对于gatk 下游分析而言,已经足够了。有时我们还会去除掉重复序列,在去除重复序列时,会根据序列的碱基质量值 ,选择一个碱基质量值总和最大的reads 作为代表序列,保留下来。扫描关注微信号,更多精彩内容等着你! 作者:生信修炼手册 链接:https://www.jianshu.com/p/cad6a359a368 来源:简书 简书著作权归作者所有,任何形式的转载都请联系作者获得授权并注明出处。
❗️在这一步之前要加上头信息:利用picard的AddOrReplaceReadGroups
这个信息随便填也行,可以直接用示例
java -jar picard.jar AddOrReplaceReadGroups \
I=input.bam \
O=output.bam \
RGID=4 \
RGLB=lib1 \
RGPL=ILLUMINA \
RGPU=unit1 \
RGSM=20
Mark Duplicates:
gatk MarkDuplicatesSpark \
-I input.bam \
-O marked_duplicates.bam
③ Base (Quality Score) Recalibration
- 这一步的目的是利用机器学习方法纠正由测序仪给出的质量分数(Base Quality Score)
- 这个误差来自:文库的制备,芯片生产,测序仪
- 校准方法:
- 收集covariate measurement from all base calls in datasets,建立统计模型。
- The initial statistics collection can be parallelized by scattering across genomic coordinates, typically by chromosome or batches of chromosomes but this can be broken down further to boost throughput if needed.
- Then the per-region statistics must be gathered into a single genome-wide model of covariation; this cannot be parallelized but it is computationally trivial, and therefore not a bottleneck.
- 利用这个模型对碱基质量进行校准 (可以并行)
- 收集covariate measurement from all base calls in datasets,建立统计模型。
四、利用Pegasus来跑GATK