人体急性呼吸道病毒感染期间口咽部的独特微生物群落景观
Unique microbial landscape in the human oropharynx during different types of acute respiratory tract infections

Article, 2023-07-24, Microbiome, [IF2023 = 15.5]
DOI:10.1186/s40168-023-01597-9
原文链接:https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-023-01597-9
第一作者:Hui Li (李辉);Xiaorong Wu (吴小蓉)
通讯作者:Tao Ding (丁涛)
主要单位:
中山医学院免疫学与微生物学系 (Department of Immunology and Microbiology, Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou 510080, China.)
中山大学热带病防治研究教育部重点实验室 (Key Laboratory of Tropical Diseases Control (Sun Yat-Sen University), Ministry of Education, Guangzhou 510080, China.)
佛山市南海区疾病预防控制中心 (Center for Disease Control and Prevention of Nanhai District, Foshan 528200, China.)
- 摘要 -
继发性细菌感染和肺炎是呼吸道病毒感染的主要死亡原因,而上呼吸道菌群失调是这一过程的关键。然而,呼吸道病毒感染期间的上呼吸道菌群失调,是否与特定病毒感染类型有关?目前尚不清楚。本研究招募了735名上呼吸道症状的门诊患者,使用多重RT-PCR确定了349名参与者的感染性病毒类型,并使用16S rRNA基因和宏基因组测序分析了他们的口咽部微生物组。结果发现,甲型流感病毒(FluA)、乙型流感病毒(FluB)、呼吸道合胞病毒(RSV)和人鼻病毒(HRV)感染形成的口咽菌群表现出三种不同的生态失调模式,韦荣氏菌属(Veillonella)被确定为任何类型呼吸道病毒感染的重要生物标志物。基于GRiD算法(Growth Rate InDex algorithm)预测细菌生长速率的结果发现,流感病毒感染后,呼吸道病原体的预测生长速度增加,而共生菌如婴儿链球菌(Streptococcus infantis)和缓症链球菌(Streptococcus mitis),可能对这些病原菌的过度生长起到定植抗性作用。这些发现对于阐明健康参与者和急性呼吸道病毒感染的呼吸道微生物群的差异和动态至关重要,有助于阐明病毒-宿主-细菌相互作用的发病机制,为未来有效预防和治疗呼吸道感染的研究提供见解。
- 引言 -
呼吸道病毒感染如流感等,易引发继发性细菌感染,是全球公共卫生的主要威胁。微生物构成了一个复杂的与人体共生的群落,与人类健康和各种疾病密切相关。病毒感染破坏了这个复杂系统所维持的微生物-宿主平衡。机体多种因素的相互作用,如病毒、细菌和宿主免疫系统,使机制的阐明变得复杂。我们仍然不清楚呼吸道病毒感染塑造的呼吸道菌群特征,以及它是否具有病毒类型特异性。
呼吸道微生物群被认为是呼吸道健康的守门员,可分为上呼吸道和下呼吸道两部分,其中前者的微生物密度明显高于后者。在上呼吸道中,口咽部是大多数呼吸道病毒感染和复制的重要部位,被认为是肺部微生物群的主要来源,并参与继发性肺炎。因此,需要基于多种病毒感染的大样本量的人体研究,来揭示不同病毒感染状态下上呼吸道菌群失调特征的异同,对其内在作用机制的挖掘,有望进一步表征这种菌群失调与下呼吸道继发性细菌感染之间的内在联系。
- 结果 -
1. 上呼吸道病毒感染参与者的人口统计学特征
Demographic characteristics of the participants with upper respiratory virus infection
从2019年3月到8月,共纳入735名流感样症状患者参与此项研究。多重RT-PCR用于FluA、FluB、HRV或RSV等呼吸道病原的鉴定,其中255例患者被确定为单独感染,94例被确定为多重感染。此外,我们招募了98名健康参与者作为对照(图1A-B)。
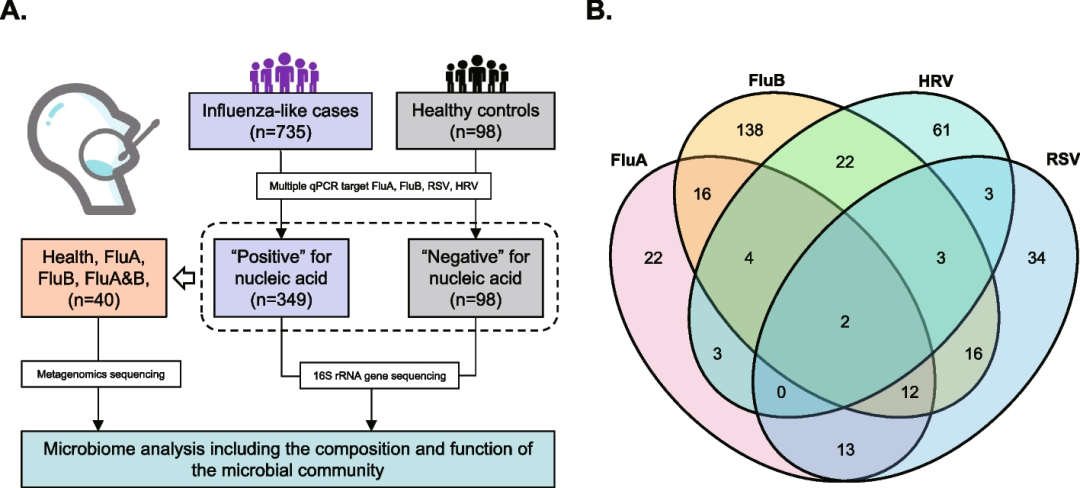
图1 研究流程及呼吸道病毒的人群分布
A. 研究队列与流程;
B. 维恩图表征不同呼吸道病毒的临床合并感染情况。
2. 呼吸道病毒感染与特征性上呼吸道菌群紊乱有关
Viral infections are associated with disorders of specific characteristics of the upper respiratory microbiota
基于Bray-Curtis距离进行PCoA分析和PERMANOVA统计,以评估不同病毒感染期间的上呼吸道(口咽)菌群的组成差异。结果显示,与健康个体相比,除RSV外,所有类型的病毒在口咽菌群组成上都具有显著的变化(图2A)。与健康个体相比,在FluA、FluB、HRV或RSV患者中,Veillonella显著富集。与健康对照组相比,任何类型病毒感染的参与者中Granulicatella的相对丰度都较少(图2B-D)。FluA感染的口咽菌群与RSV和HRV组不同,两者表现出相似的菌群特征,而FluB感染后的组成特征变化介于两者之间(图2E)。
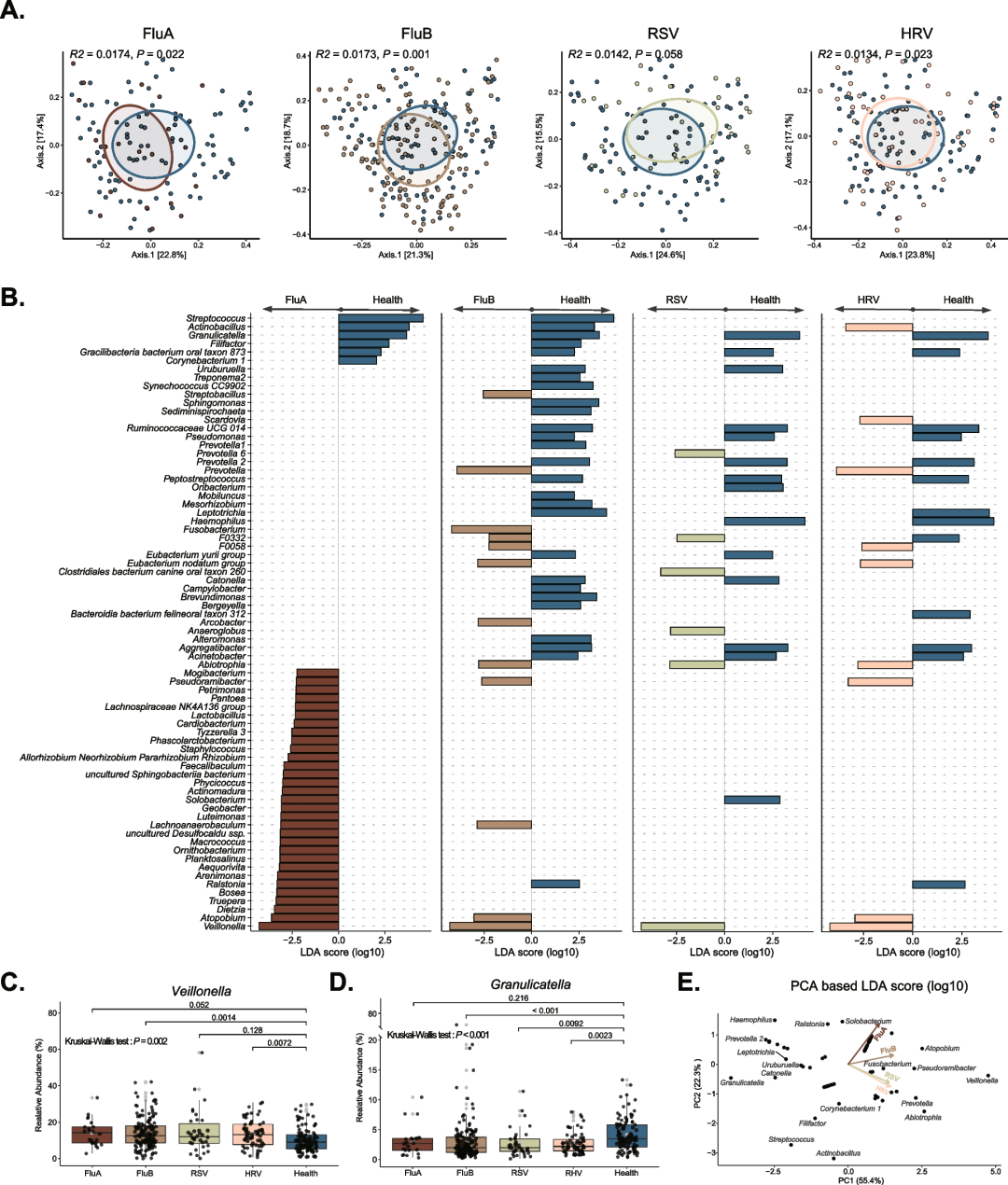
图2 上呼吸道菌群紊乱与病毒感染相关
A. 基于Bray-Curtis距离的PCoA分析,利用PERMANOVA统计病毒阳性组与健康组之间微生物组成的差异;
B. LEfSe计算病毒阳性组与健康组之间的属水平物种差异,取各组差异物种的并集用于绘制富集特征;
C, D. 病毒阳性组和健康组之间Veillonella及Granulicatella的相对丰度差异(Bonferroni校正的Wilcoxon秩和检验);
E. 基于差异物种的LDA值进行PCA分析,采用Biplot方法展示感染组(箭头)与细菌属(点)的相关性。
随机森林模型分析指出,口咽菌群能够有效区分特定病毒感染患者和健康人。此外,基于曲线下面积(AUC值)评估,我们发现随机森林模型对于FluB组(AUC=93.89%)比FluA组(AUC = 84.30%)、RSV组(AUC = 83.87%)和HRV组(AUC = 89.24%)组的区分效果更好(图3A-B)。基于特定物种的Mean Decrease Accuracy值绘制条形图,表征可以有效区分感染与非感染参与者的咽部物种差异,我们发现来自Granulicatella属的ASV能够作为FluA特异性标志物(图3C)。
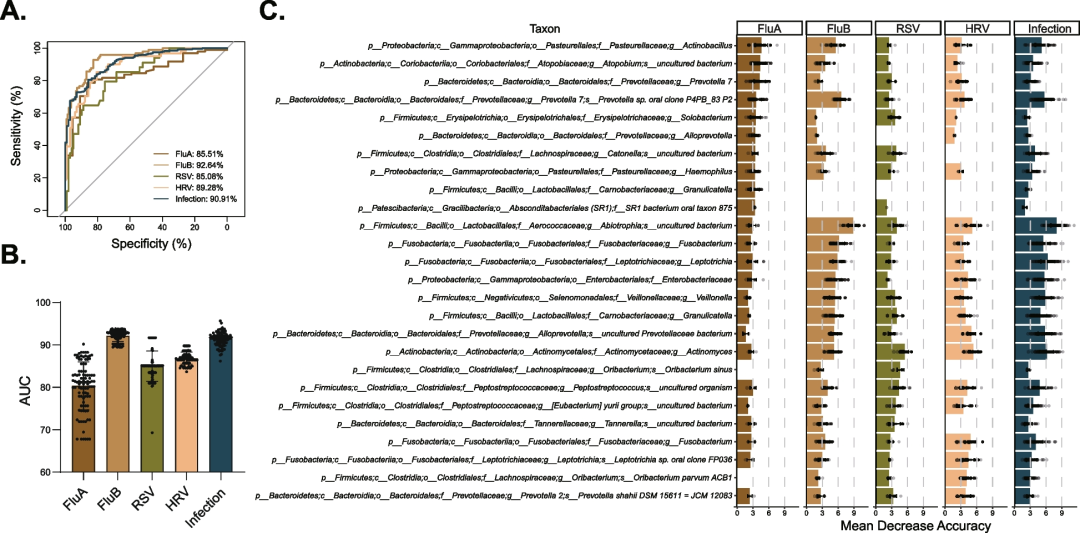
图3 随机森林模型的构建及特征细菌评估
A. 利用随机森林模型判别志愿者是否感染甲型流感病毒(FluA)、乙型流感病毒(FluB)、呼吸道合胞病毒(RSV)、鼻病毒(HRV),并计算相应的ROC曲线;
B. 利用感染样本和健康样本进行随机(n = 100)欠采样,用于随机森林模型拟合,计算AUC值评价预测效果;
C. 各分类组中Mean Decrease Accuracy值较高的Top10物种。
3. 流感病毒感染与上呼吸道微生物群多样性增加和功能改变有关
Influenza virus infection is linked with increased diversity and functional changes in the upper respiratory microbiota
通过分析不同性别、年龄、BMI和病毒感染类型相关的咽部菌群特征,发现只有FluA阳性患者的咽部菌群α多样性显著高于健康人(图4A),FluB阳性患者的β多样性与健康人存在显著差异(图4B),提示流感病毒感染与上呼吸道菌群之间的密切联系。利用宏基因组测序分析,我们发现FluA单独感染组和FluA/FluB混合感染组显示出多种微生物功能途径的富集,例如biotin biosynthesis II, L-arginine biosynthesis III(via N-acetyl-L-citrulline)等途径。同时,流感阳性患者的耐药基因多样性明显低于健康受试者。然而,无论是FluA、FluB还是混合感染,四环素耐药基因tetW含量都较高。我们进一步构建了差异通路、差异耐药基因(ARGs)和差异物种之间的相互作用网络,以表明流感感染患者与健康个体中不同的微生物特征联系(图4C),我们发现,流感感染与V. parvula的富集、多种微生物功能途径以及四环素耐药基因的富集存在潜在联系,相关机制有待进一步研究。
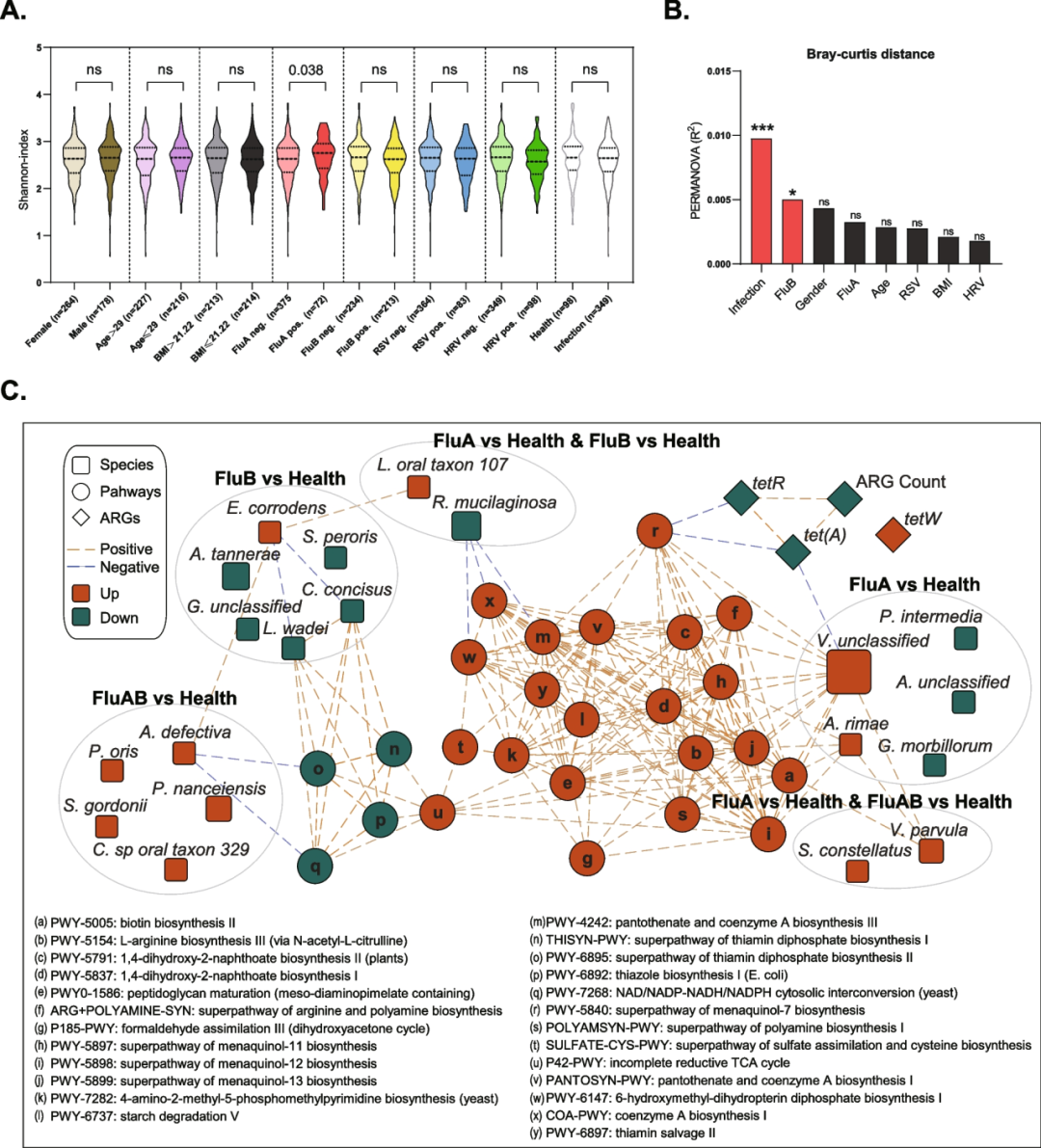
图4 宏基因组分析揭示流感病毒感染导致的菌群紊乱特征
A. 感染和健康参与者之间的咽部菌群α多样性差异;
B. 感染和健康参与者之间的咽部菌群β多样性差异;
C. 网络分析展示各分组差异通路、差异耐药基因(ARGs)和差异物种之间的Spearman相互作用网络,Cytoscape绘制关联网络。
4. 特定呼吸道病原体在流感感染者中具有较高的生长速率
Specific respiratory pathogens exhibit higher growth rates in influenza-infected individuals
基于宏基因组测序数据,我们计算了呼吸道9种潜在病原菌的生长速率(Growth Rate InDex algorithm, GRiD算法),结果表明,健康对照中致病菌的生长速度显著低于共生菌。然而,在感染个体中没有观察到共生菌对病原菌的抑制效应,这表明流感病毒感染可能创造了利于病原菌生长的环境(图5A)。同时,我们的研究结果发现了几种具有益生作用的咽部共生菌,如Streptococcus infantis、Streptococcus mitis等,这些物种的生长速度与病原菌呈显著负相关,表明它们可能通过定植抗性发挥作用(图5B)。基于Spearman构建的潜在病原菌、功能途径和耐药基因间的关联网络结果表明,T. denticola的高生长速率与formaldehyde assimilation III(dihydroxyacetone cycle)等途径的富集显著相关,T. medium的高生长速率与biotin biosynthesis II, 1,4-dihydroxy-2-naphthoate biosynthesis II途径的富集相关,而H. sputorum则与CMP-3-deoxy-D-manno-octulosonate biosynthesis I途径的富集相关(图5C)。总之,流感病毒感染患者的呼吸道病原菌生长速度显著增强,而细菌间的定植抗性、以及多种细菌途径和耐药基因可能参与了这一过程。
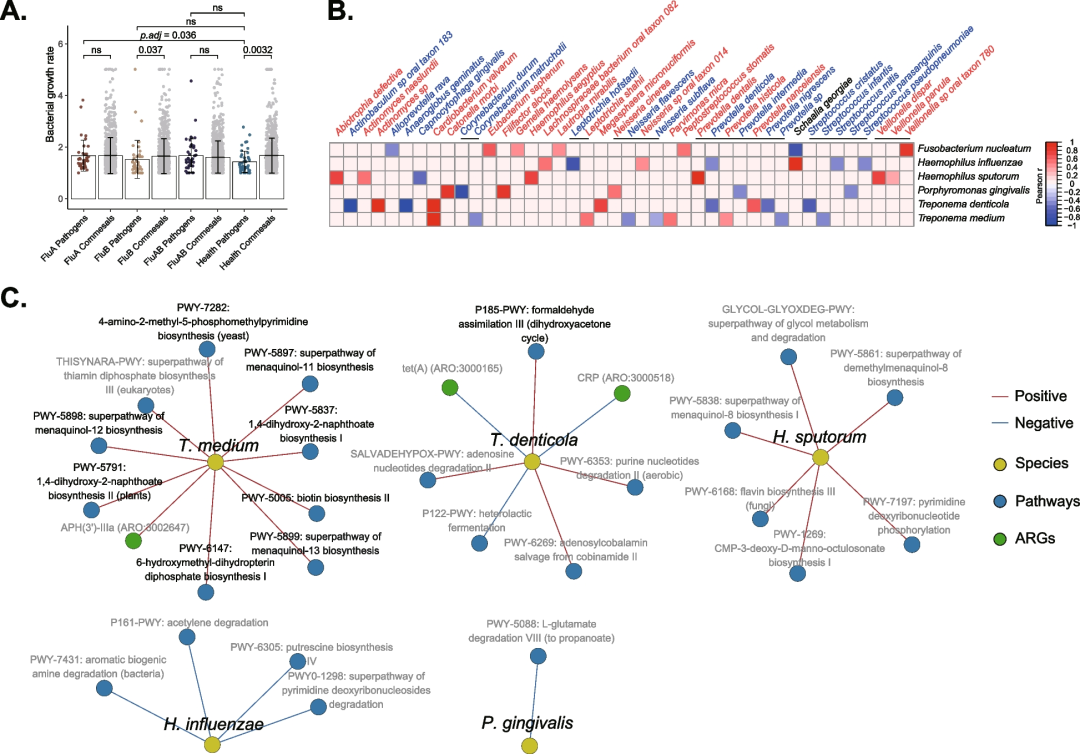
图5 流感感染患者呼吸道病原菌及共生菌的生长速率异质性
A. 利用宏基因组分析得到的潜在病原菌和共生菌的生长速率值,统计感染组与健康组之间的差异(Bonferroni校正的Wilcoxon秩和检验);
B. 基于潜在病原菌与共生菌生长速率的相关性进行分析,热图显示了具有显著相关性的对象;
C. 潜在病原菌与细菌途径或抗生素基因之间的显著性关联,Cytoscape绘制关联网络。
- 讨论 -
我们开展了一项大型队列研究,揭示了临床复杂的呼吸道病毒感染状态下的病毒特异性咽部微生态失调特征。我们发现与健康参与者相比,所有类型的病毒感染组中的Veillonella都显著富集。我们的先前研究表明,Veillonella能够从口腔迁移到肺部,并与下呼吸道的炎症和功能受损有关(Advanced Science | 中大丁涛/田国宝等揭示口腔菌群的差异化输入塑造了与健康状况相关的两种肺型)。结合本研究,我们合理推断,局部气道微生物群失调可能受到病毒感染的影响,最初发生在口腔,随后通过口-肺轴影响远端微生物群。因此,针对口咽微生物组的干预策略具有一定的应用前景。研究发现FluA、FluB、HRV和RSV感染形成了三种不同的生态失调模式,表明微生物群失调具有特定的病毒类型相关特征。根据这种病毒特异性微生物疾病的特点,我们利用机器学习模型建立了不同感染类型的微生物分类器,这些独特特征的发现有助于我们更好地了解病毒的发病机制和精准治疗。此外,我们发现甲型流感病毒感染与上呼吸道微生物组多样性的变化有关,并提出致病细菌生长速度的增加是流感病毒感染的关键过程。这些结果提示,上呼吸道微生物组扰动在连接呼吸道病毒感染和严重继发感染之间可能发挥关键作用。
- 参考文献 -
Li, H., Wu, X., Zeng, H. et al. Unique microbial landscape in the human oropharynx during different types of acute respiratory tract infections. Microbiome 11, 157 (2023). https://doi.org/10.1186/s40168-023-01597-9
- 作者介绍 -
第一作者

中山大学
中山医学院
李辉
博士后
2022年博士毕业于中山大学,获理学博士学位。从事人体微生物组与宿主免疫应答的相互作用研究,基于人群队列数据和体内外实验,探究微生物组与免疫系统的互作机制。重要研究成果以第一/共一作者发表在Microbiome、JAMA Oncology等国际知名期刊。
通讯作者

中山大学
中山医学院
丁涛
教授,博士生导师
丁涛教授团队长期从事人体微生物组与宿主健康之间的交互作用、菌群组装和决定机制的研究,欢迎对此研究方向感兴趣的博士后和研究生加入。
详情:https://zssom.sysu.edu.cn/zh-hans/teacher/396
- 相关文章 -
AS | 中大丁涛/田国宝等揭示口腔菌群的差异化输入塑造了与健康状况相关的两种肺型
MS | 中山大学丁涛/吴忠道-肠道菌群调控血吸虫病传播媒介光滑双脐螺适生性的新机制
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文