
使用宏基因组数据培养未培养微生物的机遇和挑战
Opportunities and challenges of using metagenomic data to bring uncultured microbes into cultivation
Microbiome [IF: 14.650]
DOI:https://doi.org/10.1186/s40168-022-01272-5
发表日期:2022-05-12
第一作者: Sijia Liu(刘思佳)1,2
通讯作者:Shengguo Zhao(赵圣国)([email protected])1, Jiaqi Wang(王加启)([email protected])1
合作作者: Christina D. Moon, Nan Zheng(郑楠), Sharon Huws
主要单位:
1中国农业科学院(State Key Laboratory of Animal Nutrition, Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, No. 2 Yuanmingyuan West Road, Haidian, Beijing, 100193, China)
2兰州大学(College of Pastoral Agriculture Science and Technology, Lanzhou University, Lanzhou, 730020, China)
3新西兰AgResearch草原研究中心
4英国贝尔法斯特女王大学
图文摘要
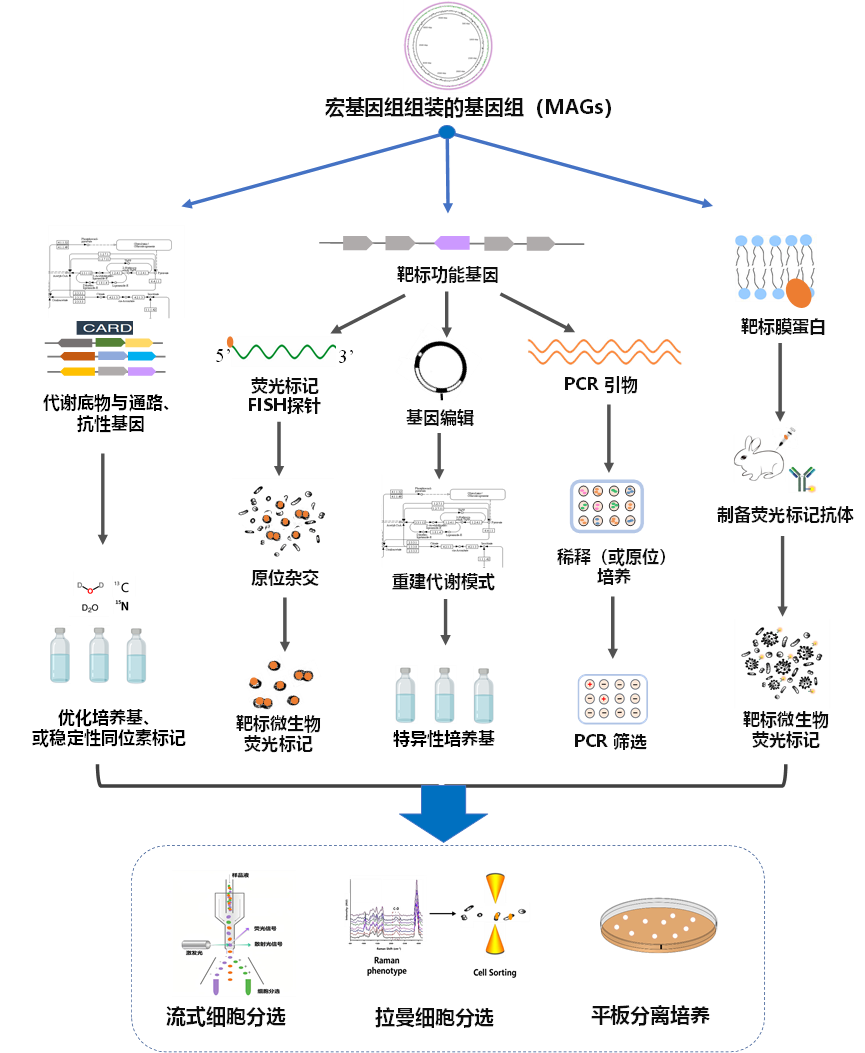
宏基因组指导未培养微生物分离培养方法
视频摘要
https://www.bilibili.com/video/BV16t4y1x7vr/
摘要
宏基因组测序技术让人们对地球上微生物的多样性有了更深入的了解,但分离培养是研究微生物的生理代谢功能并解析其生态作用的关键。目前环境中的大多数微生物仍未被分离培养,因此,分离未培养微生物是当前研究的首要任务。分离的微生物菌株可用于开发新的益生菌、生物调控剂和农业工业添加剂等。当前,宏基因组数据日益丰富,为指导靶标微生物的分离和培养带来了新的机遇。本文介绍了基于宏基因组数据分离和培养靶标微生物的新方法及应用,主要包括特定培养条件的设计、特异性抗体捕获分离、靶向基因筛选分离等技术。通过未培养微生物基因组信息预测其代谢特征和生长需求,为分离培养新的微生物并解析其功能特征提供了突破机遇。
前言
微生物是地球上已知的最早生命形式,其生存的历史可追溯到30多亿年前。微生物的多样性很高,存在于几乎所有的环境如土壤、海洋等,以及宿主体内,例如动物肠道、植物根际等,它们与宿主共同进化并与宿主的健康和相关功能紧密关联。微生物具有巨大代谢和生理多样性,在地球上几乎所有生物化学循环过程中都是必不可少的。此外,微生物与宿主相互作用在自然界中广泛存在,微生物可以帮助宿主吸收或合成机体必需的营养物质,分解代谢机体无法消化的底物,另外,微生物自身也可以作为营养来源被宿主利用。它们还可以保护宿主免受病原体和有毒物质的侵害,调节寄主的生理功能,包括免疫、发育、甚至社会行为。随着DNA测序技术的不断发展,改变了我们对地球上微生物多样性的认识,特别是细菌和古菌。同时,这些研究也揭示了大多数微生物物种仍未被培养,其中包括很多优势物种。因此,目前我们对微生物的了解,部分来自少数已被分离培养的物种,部分来自测序获得的基因组数据。虽然基于测序技术为微生物的多样性和功能提供了很多重要的新见解,但获得重要的未培养微生物的纯菌株对于直接评估其代谢和生理功能以更好地理解它们在自然环境中的生物学和生态学作用至关重要。
随着“组学”的不断发展,人们认识到培养微生物的重要性,在过去的十年里,将未被培养的微生物“暗物质”进行分离培养越来越受研究者的关注。传统的非靶向微生物培养方法和高通量培养方法如培养组学分离了许多新的和未被培养的微生物。但这种方法费时,需要消耗很多材料,并且不一定能捕获群落中重要的靶标微生物群。因此,通过靶标微生物的基因组数据,确定其独特属性以制定分离策略具有重要的应用前景。基于宏基因组测序或单细胞测序获得微生物的基因组,对靶向分离微生物具有巨大的潜力,这种方法充分挖掘并利用了未培养微生物的生物和遗传资源。本文对利用宏基因组数据指导的微生物分离培养方法的研究进展进行了综述。
未培养微生物
目前估计的全球微生物多样性仍存在很大争议,基于16S rRNA基因数据集估算全球细菌和古细菌的多样性,预测存在220-430万个原核生物可操作分类单元(OTUs;以97%相似性进行聚类)。但全球原核生物的数量远超过该数字,因为这些研究中无法获得物种的基因组和菌株表型多样性的数据。此外,真菌、原虫和其他单细胞真核生物等重要微生物类群在多样性估算中经常被忽略,但它们对微生物生态系统的功能具有重要贡献。目前,已被培养的细菌物种大多来自四个细菌门:拟杆菌门、变形菌门、厚壁菌门和放线菌门。这些细菌门在肠道微生物群落中占主导地位,这可能是由于人们对人类肠道菌群研究较多。此外,据估计,全球微生物群落中,未被培养的微生物属和门分别占81%和25%。环境中未培养的微生物物种占主导位置,具有重要的生态作用,因此深入了解它们的生物学特性对于揭示它们对生态系统的贡献是至关重要的。在过去十年中,从不同环境中分离和培养微生物的进展缓慢,因为人们对大多数微生物生长所需的复杂因素仍不了解,导致无法在实验室进行分离培养。微生物生长的自然环境通常是复杂的,并且不同微生物物种生长的最适pH值、温度、压力以及未知生长因子等参数各不相同,因此很难模拟其营养需求。此外,一些微生物生活在厌氧或其他极端环境中,这需要在实验室配置相关的专业设备。有些微生物在自然界中以休眠状态存在,导致在实验室中难以分离培养。在自然环境中,有些微生物需要交叉喂养或与宿主及其他群落成员进行相互作用。环境中一些低丰度或稀有微生物,需要进行富集后才能被分离,同时也需要其他强有力的筛选和分析方法。分离生长缓慢的细菌是一项具有挑战性的任务,因为在体外分离时,这些细菌可能无法与快速生长的细菌相竞争。
微生物培养的重要性
纯菌株是直接研究微生物功能的重要生物资源,可通过功能表型特征鉴定基因组预测的新基因,从而提高基因注释的准确性。纯菌株还可构建参考基因组,联合参考基因组、宏基因组及宏转录组数据可进一步解析微生物群落的功能。此外,纯菌株能为生物产业或健康产业提供新的生产思路和生物资源。
纯菌株可以研究微生物在特定条件下的代谢活性,揭示其未知的生理特性。这些特性无法从基因组数据中推断出来,因为基因组信息是无法确定哪些基因可以表达的,也无法确定不同基因产物之间的相互作用以及不同环境条件对基因表达的影响。有研究通过对纯菌株进行试验,发现了新的生化途径和酶促反应,而这些功能信息通过基因组数据是无法获得的。宏基因组学是解析未培养微生物强有力的工具,让科学家提出了很多新的假设,但活的纯菌株是验证这些假设的重要资源。此外,物种间的相互作用、进化原理、种群动态和致病性也只有通过纯菌株才能得到证实。微生物纯培养物还可应用于工业生产、益生菌开发等生产中。未来可收集代表性培养物构建微生物资源库,实现资源共享,用于开发新的生物技术。
微生物纯培养物可丰富参考基因组数据库和系统分类,这将进一步增加对生态系统中微生物功能的认识。目前在KEGG等基因数据库中,几乎有一半的基因功能无法确定,此外,很多注释是不准确的。通过微生物纯培养对鉴定的基因进行功能验证,将大大提高注释范围和准确性,从而进一步提高未培养微生物的可培养性。此外,微生物分类是描述微生物多样性的核心。目前,国际原核生物命名规则是基于可培养微生物进行,未培养微生物无法被正式命名。虽然为了改变这一现状,有研究者提出通过DNA序列作为微生物命名的依据,但如果没有该新物种可培养的代表菌株,则不能在分类学上进行准确的描述。此外,有研究者建议开发一个未培养原核生物的命名代码,将未培养微生物的分类整合到现有分类框架中。
有些微生物无法单独生长,因此富集培养和共培养对了解这些微生物也很有价值。富集培养是通过设计特定的生长条件,优先刺激特定微生物的生长,来提高样品中特定微生物类群的丰度。如果目标微生物的生长依赖于其他微生物,例如交叉喂养(一种微生物的代谢产物作为另一种微生物生长的底物),则需要将两种或多种微生物进行共培养。
未培养微生物分离培养的意义
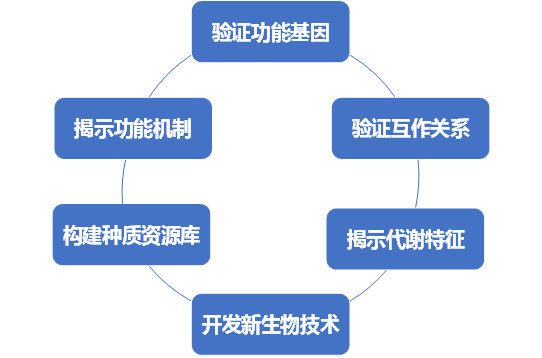
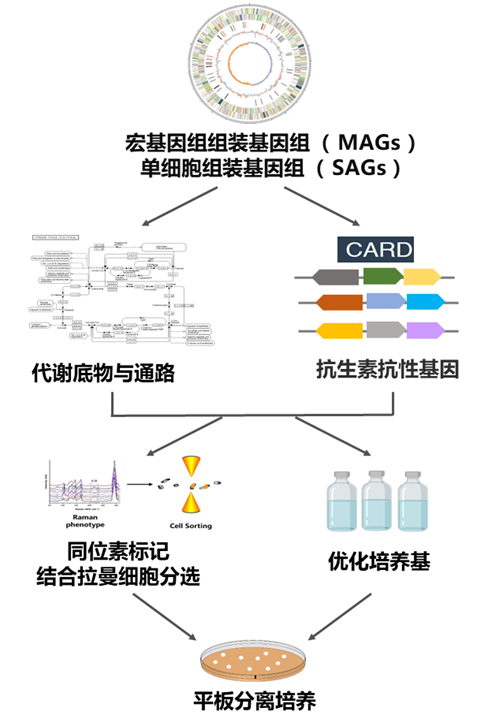
宏基因组数据重建微生物基因组及其代谢途径
宏基因组学是对微生物群落中所有基因组进行研究,通常采用鸟枪法测序对微生物进行物种分类和功能注释。宏基因组序列组装是指将每个DNA序列拼接在一起,这是基因组分析的一个关键步骤。宏基因组组装基因组(MAGs)是通过对具有相似特征的contigs进行分箱和质量控制生成的。
短读长测序技术(NGS)如Illumina测序,因其高通量和低成本而广泛应用。微生物基因组可以通过短读长测序数据进行组装,但宏基因组组装的基因组会受到重复元件的限制,而长读长测序可能克服这一问题。长读长序列可以跨越16S rRNA基因、微型倒置重复转座元件、转座子、基因复制和原噬菌体序列等常见的重复元件,显著提高MAGs的组装质量。随着测序技术的不断发展,长读长测序技术如单管长片段序列(stLFR)、PacBio和Nanopore等已得到应用。这些技术已成功从土壤、淡水湖和人类粪便等样本中重建了完整的或单个环状的基因组。虽然MAGs已经成为宏基因组数据分析流程中的标准方法,但它在16S rRNA基因、生物合成基因簇(BGCs)、潜在污染、嵌合基因、染色体或基因缺失、区分多个染色体和染色体外成分的能力上仍有局限。使用长读长测序技术和新的分箱算法可提高MAGs的质量,有时可以产生完整的环形MAG,从而提高功能注释和代谢途径重塑的效率。
除了宏基因组测序,单细胞测序是另一种获得微生物基因组信息的方法。单细胞测序是对环境样本中的单一细胞进行测序得到其基因组信息,该方法在本文中也进行了讨论,因为该方法同样可以获得未培养微生物的基因组信息。该技术已经发展成熟,目前可进行高通量测序,一次试验可得到数百个高质量的微生物单细胞基因组(SAG),对指导分离尚未培养的微生物提供基因数据。
将MAGs或SAGs预测的基因进行功能注释,重建微生物的代谢途径,对于理解微生物潜在功能至关重要,可帮助实现微生物的分离培养。宏基因组功能注释包括两个步骤:基因预测和基因注释。基因注释常用的数据库包括KEGG、EggNOG、dbCAN、RGI、GO、COG、MetaCyc、BioCyc、Brenda、Rhea、EcoCyc等。
基于公共数据库中现有的参考代谢途径,可以基于微生物蛋白质序列的同源性进行比对,预测和重建微生物代谢途径。目前重建代谢途径的方法包括:BlastKOALA、KAAS、GhostKOALA和RAST。然而,仍有许多代谢途径无法被识别,或代谢途径中的部分环节无法被识别,仅能构建出一个不完整的代谢途径。此外,这些方法不能预测参考途径中不存在的新反应或酶。为了避免出现上述情况,需要对基因间的联系有充分了解,如基因-基因间的相互作用,然后根据微生物的功能和系统发育分析进行分类。通过解析这些数据,确定微生物的营养和生长需求,指导未培养微生物分离和培养。
基于微生物的基因组信息,可以设计分离未培养微生物的方法或策略。这些方法与传统的非靶向和高通量培养方法如培养组学、原位培养、单细胞分离和iChip可相互补充。宏基因组数据分析方法和新培养方法的不断发展,为开发设计靶向分离目标种类或功能微生物的方法提供了新思路。
宏基因组数据指导的微生物分离方法
优化培养基
了解营养需求和代谢特性对未培养微生物分离和培养是至关重要的。宏基因组数据提供了微生物代谢、底物利用、氧需求、抗生素耐药性等信息,也提供了微生物与宿主相互作用信息,为微生物培养基和生长条件的设计提供了理论依据(图1)。然而,通过基因组数据了解目标微生物的生理特征并不容易,这需要深厚的代谢途径和生理学知识背景。但是,这些方法已经成功用于海洋和肠道等不同环境中分离和培养新的微生物。
图1 基于特定培养条件的设计分离靶标微生物的方法流程
Tyson等通过解析酸性矿井排水生物膜的宏基因组数据,设计改良培养基成份,成功分离到一株参与固氮的钩端螺旋体(Leptospirillum)。钩端螺旋体(Leptospirillum)基于16S rRNA基因被分为I、II、III三类。在该研究之前,已经得到了I类和II类的代表菌株,但III类的代表菌株仍未被分离培养。Tyson等人在III类钩端螺旋体(Leptospirillum)的基因组中发现nif基因操纵子,这在II类中是缺失的。作者推断,同样存在于生物膜中的II类钩端螺旋体(Leptospirillum)缺乏固氮基因。因此,作者设计了无氮培养基,成功分离出第一个III类钩端螺旋体(Leptospirillum)的代表菌株。
当摄入相同的能量时,食草性塔玛沙袋鼠(Tammar wallaby, Macropus eugenii)比反刍动物瘤胃产生的甲烷少很多,研究显示食草性塔玛沙袋鼠(Tammar wallaby, Macropus eugenii)的肠道微生物群组成独特,来自琥珀酸弧菌科(Succinovibrionaceae)的OTUs与甲烷产量相关。Pope等人利用MAG数据,部分重建了琥珀酸弧菌科(Succinovibrionaceae)WG-1的氮和碳利用途径及抗生素耐药性。预测发现WG-1的碳水化合物发酵底物为淀粉,其基因组中包括脲酶基因簇,编码运输和代谢尿素所需的所有基因(13个)。根据这些信息,作者设计了一种培养基,以淀粉和尿素作为唯一的碳源和氮源。基因组中包含一个杆菌肽抗性基因,所以在培养基中添加了杆菌肽。作者成功利用该培养基从袋鼠食糜样品中富集WG-1,并通过稀释平板法分离到纯菌株。该研究对WG-1的底物利用和培养发酵进行了详细的描述,对研究细菌与产甲烷菌的营养互作提供了基础。
宏基因组测序能鉴定新微生物生长所需的特定底物,利用该信息设计培养条件从而实现对新物种的分离培养。Lugli等人利用该方法成功从动物粪便样品中分离出新的双歧杆菌(bifidobacteria)。首先,对宏基因组数据进行组装,然后将预测的基因与糖基水解酶(GH)数据库进行比对,评估双歧杆菌可发酵的多糖。作者预测新双歧杆菌发酵碳源为阿拉伯半乳聚糖、支链淀粉、淀粉和木聚糖四种多糖。因此,作者分别利用上述碳源作为唯一碳源设计培养基,进行分离培养试验。结果培养出了13株表型不同的双歧杆菌分离株,其中2株为新的双歧杆菌。Karnachuk等人从水井深水层分离了一种嗜热螺旋体,命名为BY33,是属于Brevinematales的一个新科。首先,作者从深水层的宏基因组数据集中鉴定出了一种新的MAG。通过基因注释发现了编码利用淀粉和麦芽糖/麦芽糖糊精ABC运输系统的基因,可摄取胞外淀粉的水解产物。最后,利用添加麦芽糖和淀粉的改良螺旋体培养基进行富集和培养,成功分离出BY33。Renesto等人通过基因组分析发现,Tropheryma whipplei在氨基酸生物合成方面存在代谢缺陷。Tropheryma whipplei的基因组中缺乏9种氨基酸的合成途径(组氨酸、色氨酸、亮氨酸、精氨酸、脯氨酸、赖氨酸、蛋氨酸、半胱氨酸和天冬酰胺),表明T. whipplei需要从外部环境中获取这些氨基酸或其前体物。因此,作者设计了一种包含这些氨基酸的培养基,并成功分离出了之前培养过的3株T. whipplei,并从临床样本中分离出1株新菌。虽然该例子仅是利用了基因组序列数据,但这种方法可以应用于未培养的SAGs和MAGs数据。
解析微生物的最适生长条件也是分离培养微生物的一个重要因素。David等人报道了一种工具,可以根据微生物的基因组信息预测其最佳生长温度。作者指出,同样的原理可以应用于其他的因素,比如温度、pH值、盐浓度、渗透压或氧浓度等。这些信息对于从极端环境中分离微生物例如嗜冷菌、嗜热菌、耐高盐菌等是至关重要的。
使用抗生素
微生物对抗生素的抗性表型直接与特定的微生物类群相关联。宏基因组学和网络分析能够分析微生物群的抗生素耐药基因(ARGs)及其共现模式(图1)。因此,可以通过该方法检测和评估不同微生物分类群对抗生素耐药性特征。绘制微生物的抗生素抗性图谱,可以有针对性地分离未被培养的特定微生物类群。Rettedel等人利用该方法揭示了人类肠道菌群中16种抗生素耐药表型的分布特征。利用不同组合抗生素的培养基,作者分离了四种微生物,其中两种是属于Oscillibacter属的新物种。在Pope等人的研究中,组装的WG-1基因组中发现了杆菌肽抗性基因,作者在设计的培养基中添加杆菌肽,最后也成功分离出WG-1。因此,抗生素抗性表型在分离培养中具有重要指导意义。
稳定性同位素标记辅助拉曼活性微生物细胞分选(RACS)
另一种分离新微生物的方法是利用稳定性同位素探针标记的拉曼活性微生物细胞分选,该方法能够获得可培养的活细胞(图1)。通过宏基因组数据可获得微生物的代谢、底物利用、氧需求、抗生素耐药性等性状的信息,根据这些信息可针对靶标微生物设计培养基并创造其最适的生长条件。已有研究证明,拉曼显微镜技术结合氘同位素探针(DIP)可以在特定的培养基中识别具有代谢活性的靶标细菌。因此,可以通过基于MAGs或SAGs信息设计特定的培养基,在使用稳定性同位素氘标记的重水(D2O)中培养靶标细菌。D2O是一种有效的DIP探针,它的C-D波段在拉曼光谱检测时会在2000-2300cm-1产生偏移。当微生物样品在含有特定代谢底物和D2O的培养基中培养时,具有代谢活性的细菌会在单细胞拉曼光谱中产生C-D波段偏移,基于单细胞拉曼光谱,利用RACS便可分离具有代谢活性的靶标微生物。
Yi等人使用RACS技术从人体肠道微生物群中原位检测具有代谢活性的耐药菌群。取粪便样品在添加40% D2O (v/v)和抗生素(阿莫西林、头孢氨苄、氟苯尼考、四环素和万古霉素)的PBS中培养24小时(37℃)。他们成功地分选出六种微生物类群,包括两种阿莫西林耐药菌(AmoxR)、两种头孢菌素耐药菌(CephR)和两种头孢菌素敏感菌(CephS)。Kang等人利用这种方法对小鼠肠道中分解粘蛋白的细菌进行分选。将小鼠结肠内容物悬浮于含50% D2O的PBS和粘蛋白中,厌氧孵育后利用RACS分选出一类可降解粘蛋白的菌群,其中包括Muribaculaceae科的多个新菌。
只要微生物细胞在拉曼光谱中有特定的峰,RACS平台就可以通过相应的参数对这一类细胞进行分类。例如,Yun等人利用13C和15N从口腔分离出五种不同类型的细菌。这些研究表明拉曼活细胞分选技术在靶向识别和分离特定代谢过程中的关键微生物方面具有潜力。
反向基因组(特异性抗体)指导微生物分离
Cross等人介绍了一种基于基因组信息设计和制备特异性抗体的分离方法,通过荧光标记的特异性抗体结合细胞分选技术可从复杂微生物群落中分离新的微生物,这一方法被称为反向基因组学(reverse genomics)。该方法使用特异性抗体,能够有效地标记靶细胞,同时保留其培养活性(图2)。该方法流程如下,根据未培养的微生物基因组数据的MAGs或SAGs信息确定感兴趣的菌株,预测靶标微生物基因编码的膜蛋白与细胞外的抗原表位,合成抗原蛋白后制备特异性抗体,然后用荧光标记抗体。被标记的抗体与靶标微生物的样本结合,利用荧光细胞分选技术从样品中分离被荧光标记的靶标微生物细胞。该方法可使靶细胞在整个分选过程中保持活性,可用于后续的培养和功能验证等试验。
图2 基于反向基因组学的方法分离靶标微生物的方法流程
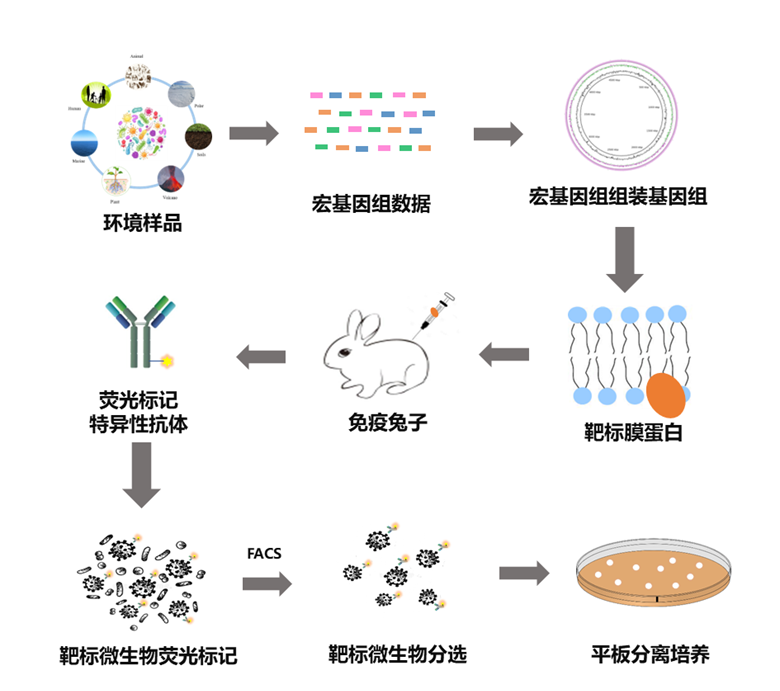
Cross等人利用该方法从人唾液样本中分离培养了Saccharibacteria (TM7)的三个不同物种。作者利用唯一的TM7基因组,基于IMG数据库(https://img.jgi.doe.gov)注释编码膜蛋白的基因。利用TMHMM v.2.0分析锚定跨膜螺旋结构域,排除假设的和短于100个氨基酸的蛋白。剩余序列与从人类口腔微生物组数据库中(http://www.homd.org/)下载的所有口腔细菌的蛋白质组数据进行比对分析。随后分析候选靶标序列跨膜结构域的数量、预测的胞外结构域的数量、大小与丰度以及与免疫相关的实验数据。排在首位的是PBP2,PBP2的高分辨率晶体结构和抗PBP2抗体与致病菌细胞表面结合的研究均已被报道。利用ABCpred59、BepiPred60、IEBD Analysis Resource (http://tools.immuneepitope.org/bcell)和Bio-Synthesis peptide design tools (https://www.biosyn.com)对蛋白序列进行抗原区域和亲水性分析。抗原肽通过商业途径生物合成,在C端(PBP2)添加半胱氨酸,纯化后注射于兔体内。HiTrap Protein A HP抗体纯化柱(GE Bio-Sciences)纯化IgG。Alexa Fluor 488 Antibody labeling Kit (A20181)试剂盒对IgG荧光标记。荧光标记的抗体与样品孵育后,利用流式细胞仪分选被标记的靶标微生物,随后通过平板分离培养靶标微生物。为了测试该方法的适用性,作者选择了一个没有3D结构和抗原-抗体试验数据的蛋白质CpsC。作者在口腔细菌基因组数据库中没有发现与CpsC同源性超过30%的蛋白。Cross等人首次分离培养了人口腔样本中丰度较低的SR1/Absconditabbacteria。反向基因组学分离微生物的方法对分离未培养的感兴趣的微生物特别是生长缓慢、丰度低和稀有的物种具有很大的应用潜力。
靶向功能基因分离策略
宏基因组数据可以检测功能基因的变异、低丰度和稀有物种,用以分离携带靶标基因的微生物。Ma等人描述了一种基于宏基因组测序数据分离培养携带靶标基因微生物的方法。该方法使用带有纳米孔的微流体装置进行,该装置可将每个单菌隔开独立生长,主要包括两个步骤:首先确定靶标微生物的培养条件然后进行分离。将样品在生长介质中稀释使在微流控芯片的每个孔中最多只有一个细胞。为了避免一个孔中出现多个细胞,需保证一定数量孔是空的。培养一段时间后,微流控芯片可将每个菌落分成2份,其中1份用于对靶标基因进行PCR,以确定靶标微生物。将样品置于芯片上进行培养,然后通过PCR筛选,确定含有靶标微生物的孔,从芯片的另一份对应的孔中取出样品进行富集培养(图3)。Ma等人利用该方法分离培养了来自人盲肠中来自瘤胃球菌科(Ruminococcaceae)的一个新属代表菌。这种方法克服了不同微生物生长动力学的偏差,试验装置体积小可节约试验材料,并可置于厌氧箱分离厌氧菌,比如肠道菌群。微体积培养的难点是气体基质的提供。但是,最近出现的微液滴供氧新方法或许可以应用于微体积培养。
图3 基于靶标基因靶向分离微生物方法流程
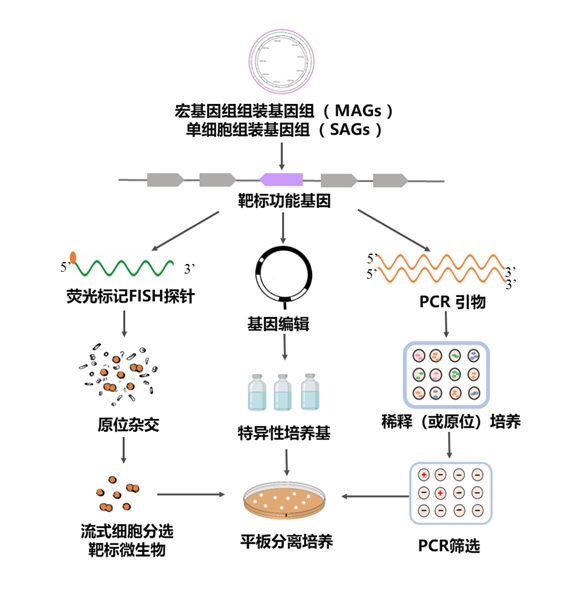
荧光原位杂交(FISH)标记的样品可以利用荧光激活细胞分选(FACS)进行分类以富集被标记的细胞,因此可利用宏基因组序列信息对目标微生物设计探针,从而实现目标微生物的富集分离培养。然而,标准的FISH程序需要对样品进行固定,使微生物失去活性无法进行分离培养。有研究通过修改FISH程序,去除了固定的步骤,改进后的程序可保留微生物的活性,用于后续分离培养试验。Tan等人通过宏基因组测序数据设计FISH探针,用于分离污水处理厂样本中的微生物。探针以V6的高可变区域序列为模板进行设计,长度33bp,通过设计的探针和FISH–FACS成功的富集了一个鞘脂杆菌目(Sphingobacteriales)的新细菌分类单元(UPWRP_1)。基于高可变区域序列信息设计的探针可鉴定到菌株水平,结合FISH活细胞分选,可以分离培养靶标微生物。
一些抗生素通过直接与细菌表面结构结合发挥作用,因此可将其作为细菌标记探针用于分离特定种类的微生物。该方法已分别在体外和体内被应用于鉴别不同的细菌种类。
此外,基因组编辑技术也可用于分离新的靶标微生物。基于MAGs序列信息对特定基因进行编辑,改造靶标微生物基因组,结合含有选择性抗生素或底物的培养基,分离编辑后的靶标微生物(图3)。Rubin等人通过编辑微生物群落中特定微生物的基因组并对其进行分离,从而验证了该方法的适用性。作者利用VcDART转座子设计了缺失pyrF基因的克雷伯菌(Klebsiella michiganensis)和假单胞菌(Pseudomonas simiae)突变株。突变株包含携带两种抗生素抗性基因的转座子,分别对链霉素、大幕霉素和青霉素耐药。通过该方法,作者成功富集了目标微生物(丰度达99%)。此外,对靶标微生物的乳糖同化基因进行编辑,并在生长培养基中添加乳糖作为唯一碳源,成功富集了靶标微生物使其丰度达到95%。基因编辑技术指导的富集方法也具有很高的特异性,可以区分同一物种中的不同菌株,例如大肠杆菌的亚群2和3。
挑战与局限性
虽然已经有许多研究利用宏基因组数据成功地分离和培养了靶标微生物,但想要分离更多的微生物,仍存在一些挑战和局限性。反向基因组(特异性抗体)为靶向分离培养微生物提供了一个新机会,但该方法仍存在一些局限性。首先,有些微生物无法确定合适的胞外抗原表位,可根据亲缘关系相近物种的同源基因进行预测,但该方法仅对部分微生物有效。其次,如果目标膜蛋白的表达水平低则无法获得其抗体,可通过基因表达和蛋白质组学数据鉴定高表达量的膜蛋白。最后,靶标抗原表位如果存在翻译后修饰如糖基化,可能会阻碍抗体的识别。为了避免上述问题,进行试验时可以选择多个抗原表位,或者使用整个蛋白结构域,但可能会降低特异性。
利用细胞分选方法分离靶标厌氧微生物更具有技术挑战。由于细胞分选仪器较大,很难放入厌氧箱,因此,可开发在厌氧条件下使用的荧光细胞分选仪器。分离到靶标微生物后,需确定其合适的生长介质和培养条件。但是,微生物栖息的自然生态环境组分是复杂的,甚至有些成份是未知的,有些微生物单靠基因组序列不能提供足够的信息来准确地确定其生长所需的所有必要条件。例如,Lavy等人根据海绵杆菌(Poribacteria sp.) WGA-4E的基因组数据设计了一种培养基。虽然根据基因组数据预测了该细菌门的代谢特性,比如以尿素为氮源,能同化硫酸盐,但根据这一信息设计的特定培养基并没有捕获到靶标菌株。海绵杆菌(Poribacteria)菌的生长除了上述所需的营养成分,可能还需要某些未知的信号分子。将宏基因组学方法与培养组学、原位培养、单细胞分离、析因试错法等相结合,将提高成功分离培养靶标微生物的机会。
基于基因组数据设计目标微生物的培养策略,很大程度上依赖于提取的DNA质量,但可能存在细胞裂解、DNA提取不完全或DNA降解等问题,这可能会影响某些物种在宏基因组数据中的代表性。单细胞基因组学是重建靶标微生物基因组的另一种方法,但这需要更专业的细胞分离、基因组扩增、测序和分析技术。
结论
DNA测序技术的快速发展揭示了自然界中存在大量尚未培养的微生物。目前面临的挑战是利用这些数据指导未培养微生物的分离,探索微生物在其自然栖息地的生态特征。宏基因组测序不仅揭示了微生物组的种类和功能特征,还为分离和培养微生物提供了新的思路和策略,具有很大的应用潜力。随着长读长DNA测序技术的发展、测序深度的增加和生物信息学方法的更新,MAGs的质量不断提高,使基因注释和代谢途径预测更加准确。基于这些数据设计特定培养基、特异性抗体和靶基因或探针的培养方法,从不同环境中分离出多种微生物。通过宏基因组数据指导微生物的分离方法,大大增加了成功培养靶标微生物的机会。然而,靶向分离和培养的过程往往是复杂的,需要深入解析基因组数据来预测微生物的培养条件,分离或分选过程涉及的先进仪器和技术,有些微生物实验室不具备。靶向分离培养方法可以节约目标微生物的分离时间。目前面临的挑战是如何利用丰富的宏基因组数据实现高通量靶向培养。结合一些先进的新培养方法,将有助于实现对未培养微生物分离培养的新突破。
通讯作者

赵圣国,1984年生,副研究员,中国农业科学院北京畜牧兽医研究所。2012年获中国农业科学院博士学位,2011-2012年在澳大利亚CSIRO Livestock Industries做联合培养博士生,2012-2014年在中国科学院微生物研究所博士后工作,2014年至今在中国农业科学院北京畜牧兽医研究所工作。主要从事动物消化道(瘤胃)微生物组学研究,揭示营养素、微生物和动物机体之间的互作关系。主持国家自然科学基金等国家级项目或子课题2项,获神农中华农业科技奖等科技奖励3项,入选农业农村部“杰出青年农业科学家”。以第一或通讯作者(含共同)发表论文67篇,其中在Microbiome、Applied and Environmental Microbiology、Frontiers in Microbiology、Journal of Dairy Science等期刊发表SCI论文37篇,出版《益生菌与动物营养——生产、效果和规范》书籍。兼任《Animal》和《生物技术通报》杂志编委,国际学术组织Metaproteomics Initiative和Anaerobic Fungi Network专家组委员。

王加启,研究员,中国农业科学院北京畜牧兽医研究所奶业创新团队首席专家,农业农村部食物与营养发展研究所所长,全国农业科研杰出人才。主要从事奶牛营养与牛奶质量安全研究。获得国家科技进步奖二等奖3项,发表SCI论文百余篇,担任《Journal of Dairy Science》编委。
Reference
Sijia Liu, Christina D. Moon, Nan Zheng, Sharon Huws, Shengguo Zhao, Jiaqi Wang. Opportunities and challenges of using metagenomic data to bring uncultured microbes into cultivation. Microbiome (2022). https://doi.org/10.1186/s40168-022-01272-5
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
点击阅读原文,跳转最新文章目录阅读