点击关注,桓峰基因
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
120篇原创内容
公众号
桓峰基因公众号推出基于基因组变异数据生信分析教程并配有视频在线教程,目前整理出来的教程目录如下:
DNA 1. Germline Mutation Vs. Somatic Mutation 傻傻分不清楚
DNA 2. SCI 文章中基因组变异分析神器之 maftools
DNA 3. SCI 文章中基因组变异分析神器之 maftools
DNA 4. SCI 文章中基因组的突变信号(maftools)
DNA 5. 基因组变异文件VCF格式详解
DNA 6. 基因组变异之绘制精美瀑布图(ComplexHeatmap)
DNA 7. 基因组拷贝数变异分析及可视化 (GISTIC2.0)
DNA 8. 癌症的突变异质性及寻找新的癌症相关基因(MutSigCV)
前 言
目前国际主要基因组项目旨在建立一个全面的、与癌症的发生和发展有关的所有基因目录。这些研究包括对匹配的正常肿瘤样本进行排序,然后进行数学分析,以确定那些突变发生频率高于预期的随机基因。在这里,我们描述了癌症基因组研究的一个基本问题:随着样本量的增加,由目前的分析方法产生的假定重要的基因列表迅速增加到数百个。该列表包括许多不可信的基因(如编码嗅觉受体和肌肉蛋白肌肽的基因),表明大量的假阳性发现掩盖了真正的驱动事件。我们表明,这一问题主要源于突变异质性,并提供了一种新的分析方法MutSigCV来解决这一问题。将MutSigCV应用于3083对正常肿瘤的外显子组序列,发现癌症类型中突变频率和频谱的异常变化,揭示了突变过程和疾病病因,以及全基因组的突变频率,这与DNA复制时间和转录活性密切相关。通过将突变异质性纳入分析,MutSigCV能够消除大多数明显的人工发现,并使识别真正与癌症相关的基因成为可能.

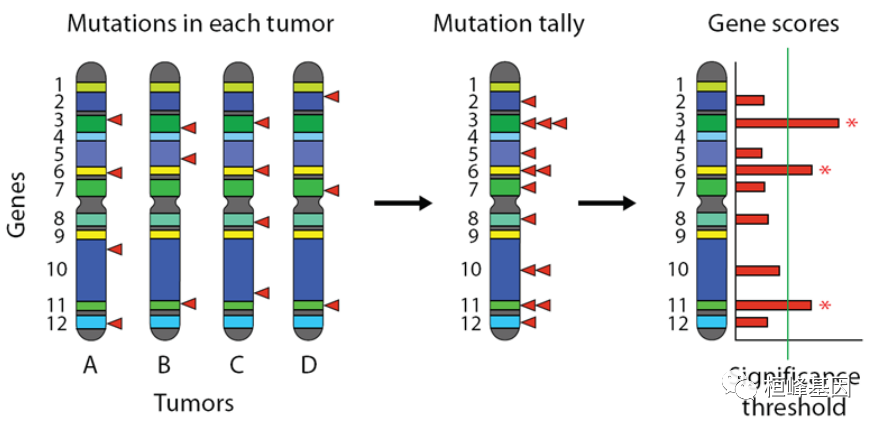
下图说明了整个概念。左边是一组基因组(或外显子组),每个都来自不同癌症患者的肿瘤细胞测序。基因用彩色条纹表示,体细胞突变用红色三角形表示。首先,将肿瘤聚集在一起,统计突变,然后计算每个基因的得分和p值。选择显著性阈值来控制虚假发现率(FDR),超过该阈值的基因被报告为显著突变。
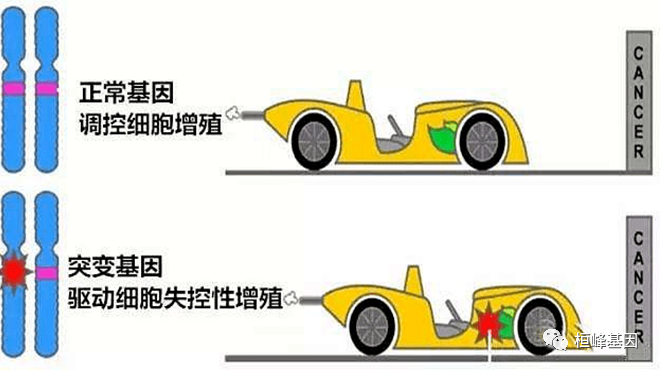
突变的分类方法有很多种,按照其是否会导致癌症进展,可以分为驱动突变(driver mutation)和乘客突变(passenger mutation)。前者在肿瘤细胞中具有选择性生长优势的突变,后者对肿瘤细胞的选择性生长优势无直接或间接影响的突变。
软件下载安装
下载及安装包括两部分,一部分是需要先安装matlab,如果您的已经配置完成,那请忽略;一部分是MutSigCV安装。
- 安装 matlab
在软件中指出使用的matlab版本号:R2016a
$le readme.txt
MATLAB Compiler
1. Prerequisites for Deployment
. Verify the MATLAB Runtime is installed and ensure you
have installed version 9.0.1 (R2016a).
官网下载链接:
https://ssd.mathworks.cn/supportfiles/downloads/R2016a/deployment_files/R2016a/installers/glnxa64/MCR_R2016a_glnxa64_installer.zip
解压安装
cd ~/software/
mkdir MatlabMCR
cd MatlabMCR
wget -c http://ssd.mathworks.com/supportfiles/downloads/R2016a/deployment_files/R2016a/installers/glnxa64/MCR_R2016a_glnxa64_installer.zip
unzip MCR_R2016a_glnxa64_installer.zip
./install
安装成功标记,如下:
(Jun 11, 2022 18:35:32) Exiting with status 0
(Jun 11, 2022 18:35:32) End - Successful.
设置环境变量
安装过程需要选择一个有权限的安装路径,安装好了之后可能要手动赋值一个变量:
LD_LIBRARY_PATH=/home/data/t030339/software/MutSigCV/MatlabMCR/v901//runtime/glnxa64:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//bin/glnxa64:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//sys/os/glnxa64:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//sys/opengl/lib/glnxa64
2. 安装 MutSigCV
先下载,之后解压即可完成,这个软件包使用 matlab 开发的,所以安装很轻松,就是注意一下上面安装 matlab 时 一些细节可以了!
cd ~/software
wget -c https://software.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/MutSigCV_1.41.zip
unzip MutSigCV_1.41.zip
cd MutSigCV_1.41
下载相应的数据文件。利用wget 下载更方便,加参数 -c 防止断掉,可以续传,这个非常重要。
wget -c http://www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/reference_files/gene.covariates.txt
wget -c http://www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/reference_files/exome_full192.coverage.zip
wget -c http://www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/reference_files/mutation_type_dictionary_file.txt
wget -c http://www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/reference_files/chr_files_hg19.zip
wget -c https://software.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/MutSigCV_example_data.1.0.1.zip
wget -c https://software.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/LUSC.MutSigCV.input.data.v1.0.zip
实例操作
1. 数据读取
MutSigCV 输入文件有 5 个,一个自己的数据,格式为.maf文件,其他四个都有软件自带的数据库文件!我们看看每个文件的格式。软件里的例子来之 2013年发表在 nature 上的一篇文章,癌症的突变异质性和寻找新的癌症相关基因。

a. LUSC.maf
我们只需要准备自己的.maf文件即可,格式如下:
i. “gene” 列:突变所在的基因的名字 (也可以称为 “Hugo_Symbol”);
ii. “patient” 列:突变所在的病人的名字 (也可以被称为 “Tumor_Sample_Barcode”);
3. “effect” 列:突变在这个基因上所产生的作用类型:分为 “nonsilent” (蛋白质序列改变或可变剪切), “silent” (同义突变),或"noncoding" (内含子区或UTR区)
4. “categ” ’列:突变分类. MutSigCV依据突变所在的DNA序列将突变分成了7类,对于每一种分类,有不同的风险值. 如果用户不知道每一行的categ类型,从版本1.3开始程序可以自动计算,只需要用户提供Variant_Classification, Reference_Allele, and Tumor_Seq_Allele1+2这4列的信息即可。
1. CpG transitions
2. CpG transversions
3. C:G transitions
4. C:G transversions
5. A:T transitions
6. A:T transversions
7. null+indel mutations
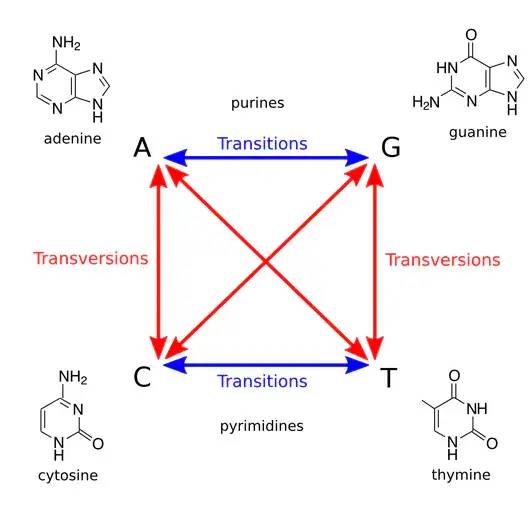
转换(transitions)和颠换(transversions)
转换:嘌呤和嘌呤之间的替换,或嘧啶和嘧啶之间的替换。
颠换:嘌呤和嘧啶之间的替换
需要注意的是,与MutSig捆绑在一起的协变量文件(gene.covariates.txt和exome_full192.coverage.txt)具有非标准的基因名称(non Hugo_Symbols)。MAF中的Hugo_Symbols与协变量文件中的非Hugo_symbols之间的差异导致MutSig程序忽略此类基因。可以用 R包 maftools 的 prepareMutSig 函数来进行纠正,获得校正后的文件。
library(maftools)
#Prepare MAF file for MutSigCV analysis
lusc<-read.maf("LUSC.maf")
lusc.mutsig.corrected = prepareMutSig(maf = lusc)
lusc_sig = read.maf(maf = lusc.mutsig.corrected)
b. exome_full192.coverage.txt
gene"列: 基因名, 与突变文件的基因名列对应
“effect"列: 分类为"silent”, “nonsilent”, or “noncoding”
-
"categ"列: 与突变文件一致
-
number of sequenced bases for patient#1 in this gene and effect/categ bin
-
number of sequenced bases for patient#2 in this gene and effect/categ bin
$ le ../exome_full192.coverage.txt
gene effect categ coverage
A1BG noncoding A(A->C)A 12
A1BG noncoding A(A->C)C 14
A1BG noncoding A(A->C)G 15
A1BG noncoding A(A->C)T 9
A1BG noncoding A(A->G)A 12
c. gene.covariates.txt
这个表格列出了每个基因的基因组参数。它们被称为协变量,因为它们与突变率共变。它们将被用来计算“协变量空间”中成对基因之间的距离,以确定每个基因的最近邻居,以便汇集关于局部背景突变率(BMR)的信息。
$ le ../gene.covariates.txt
gene expr reptime hic
A1BG 1621097 406 -25
A1CF 113129 613 19
A2BP1 2474 1138 -47
A2M 348389 364 17
A2ML1 300254 407 12
d. mutation_type_dictionary_file.txt
当MAF文件没有effect列时是必须的,另外4个文件是当我们只有MAF文件的时候需要的,建议也同时下载。
Variant_Classification effect
Silent silent
Synonymous silent
Missense nonsilent
Missense_Mutation nonsilent
Nonsense null
e. chr_files_hg19
这是一个文件夹按照染色体的生成的参考基因组序列文件,打开看看,其实就是核酸序列,如下:
$ ll ../chr_files_hg19 | le
chr10.txt*
chr11_gl000202_random.txt*
chr11.txt*
chr12.txt*
chr13.txt*
2. 实际操作
准备好所有数据就可以运行了,这个例子中的数据还算很快的,大概2-3钟左右吧!
../MutSigCV_1.41/run_MutSigCV.sh ~/software/MutSigCV/MatlabMCR/v901/ LUSC.maf ../exome_full192.coverage.txt ../gene.covariates.txt LUSC_mutsig ../mutation_type_dictionary_file.txt ../chr_files_hg19
###运行过程
------------------------------------------
Setting up environment variables
---
LD_LIBRARY_PATH is .:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//runtime/glnxa64:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//bin/glnxa64:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//sys/os/glnxa64:/home/data/t030339/software/MutSigCV/MatlabMCR/v901//sys/opengl/lib/glnxa64
======================================
MutSigCV
v1.4
(c) Mike Lawrence and Gaddy Getz
Broad Institute of MIT and Harvard
======================================
MutSigCV: PREPROCESS
--------------------
Loading mutation_file...
Loading coverage file...
Processing mutation "effect"...
NOTE: This version now ignores "is_coding" and "is_silent".
Requires Variant_Classification/type column and mutation_type_dictionary so we can assign nulls.
Processing mutation "categ"...
"categ" of mutation_file does not match coverage_file. Ignoring it.
NOTE: unable to perform category discovery, because no chr_files available.Will use two categories: missense and null+indel.
Collapsing coverage...
Writing preprocessed files.
[save_struct] 23/47 24/47 25/47 26/47 27/47 28/47 29/47 30/47 31/47 32/47 33/47 34/47 35/47 36/47 37/47 38/47 39/47 40/47 41/47 42/47 43/47 44/47 45/47 46/47 47/47 [collapse] [write]
MutSig_preprocess finished.
MutSigCV: RUN
-------------
Loading mutation_file...
NOTE: Both "gene" and "Hugo_Symbol" are present in mutation_file. Using "gene".
NOTE: Both "patient" and "Tumor_Sample_Barcode" are present in mutation_file. Using "patient".
Loading coverage file...
Loading covariate file...
NOTE: 4/16885 gene names could not be mapped to coverage information. Excluding them.
NOTE: Trimming "-Tumor" from patient names.
NOTE: Converting "-" to "_" in patient names.
Building n and N tables...
Processing covariates...
Finding bagels... 1000/18862 2000/18862 3000/18862 4000/18862 5000/18862 6000/18862 7000/18862 8000/18862 9000/18862 10000/18862 11000/18862 12000/18862 13000/18862 14000/18862 15000/18862 16000/18862 17000/18862 18000/18862
Expanding to (x,X)_gcp...
Calculating p-value using 2D Projection method... 1000/18862 2000/18862 3000/18862 4000/18862 5000/18862 6000/18862 7000/18862 8000/18862 9000/18862 10000/18862 11000/18862 12000/18862 13000/18862 14000/18862 15000/18862 16000/18862 17000/18862 18000/18862
Done. Wrote results to LUSC_mutsig.sig_genes.txt
结果解析
生成了4个结果文件,下面重要的文件都已经加一定的注释,方便大家理解,如下:
LUSC_mutsig.categs.txt
LUSC_mutsig.coverage.txt
LUSC_mutsig.mutations.txt
LUSC_mutsig.sig_genes.txt
a. LUSC_mutsig.categs.txt
left from change right autoname name type
ACGT AC in ACGT missense missense point
ACGT AC in ACGT null+indel null+indel non-point
b. LUSC_mutsig.coverage.txt
覆盖表格告诉我们有多少核苷酸被测序到足够的深度以进行突变调用。
gene effect categ coverage
A1BG noncoding missense 3309
A1BG noncoding null+indel 3309
A1BG nonsilent missense 3438
A1BG nonsilent null+indel 3438
A1BG silent missense 1116
c. LUSC_mutsig.mutations.txt
Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome Start_position End_position Strand Variant_Classification Variant_Type Reference_Allele Tumor_Seq_Allele1 Tumor_Seq_Allele2 dbSNP_RS dbSNP_Val_Status Tumor_Sample_Barcode Matched_Norm_Sample_Barcode Match_Norm_Seq_Allele1 Match_Norm_Seq_Allele2 Tumor_Validation_Allele1 Tumor_Validation_Allele2 Match_Norm_Validation_Allele1 Match_Norm_Validation_Allele2 Verification_Status Validation_Status Mutation_Status Sequencing_Phase Sequence_Source Validation_Method Score BAM_file Sequencer Genome_Change Annotation_Transcript Transcript_Strand Transcript_Exon Transcript_Position cDNA_Change Codon_Change Protein_Change gene patient effect chr start ref_allele categ
PHTF1 10745 broad.mit.edu 37 1 114242392 114242393 + Frame_Shift_Ins INS - T T LUSC-18-3406-Tumor LUSC-18-3406-Normal Phase_I Unspecified Illumina GAIIx g.chr1:114242392_114242393insT uc009wgp.1 - 16 2527_2528 c.2075_2076insA c.(2074-2076)AAGfs p.K692fs PHTF1 LUSC-18-3406-Tumor null 1 114242392 - null+indel
d. LUSC_mutsig.sig_genes.txt
****该文件即为MutSigCV 的结果报告。MutSig输出分析中的“ nnei”,“ x”和“ X”值是算法计算得到的基因背景突变率。我们重点关注的是显著性的 p 值和 q 值,后者为校正过的 p 值。****该文件中包含了Driver Gene,从P值由小到大排列。
gene expr reptime hic N_nonsilent N_silent N_noncoding n_nonsilent n_silent n_noncoding nnei x X p q
KEAP1 1996822 207 30 780570 225675 327627 24 0 1 36 117 41857491 0 0
结果展示
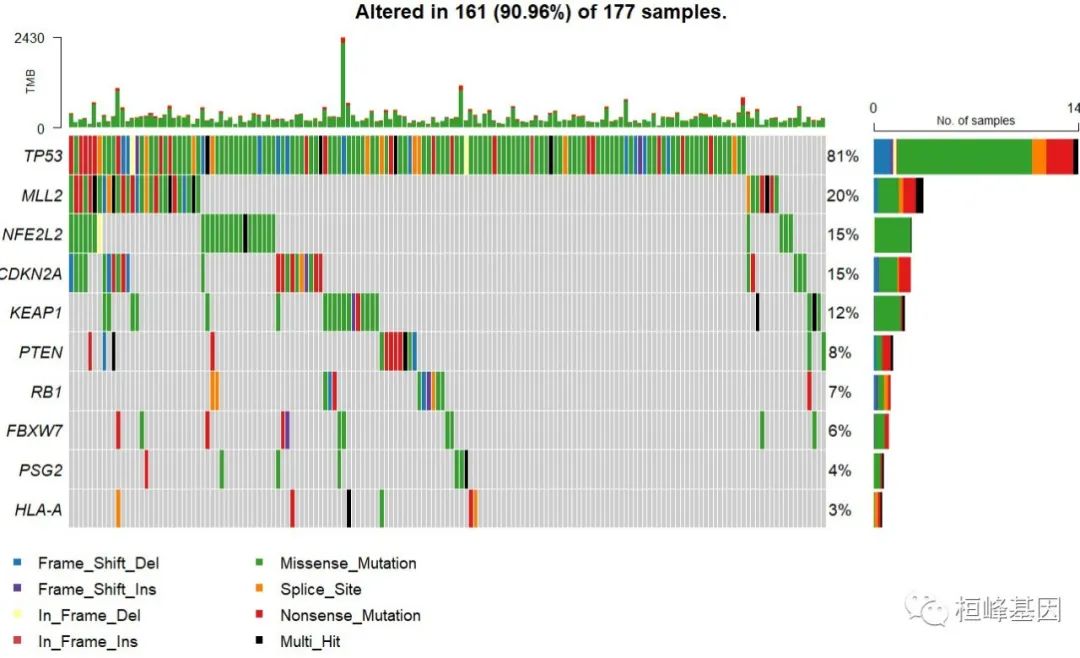
根据最后的结果报告,筛选 q<0.05 的基因,通过使用R包maftools绘制瀑布图,如下:
library(maftools)
lusc = read.maf(maf ="LUSC_mutsig.mutations.txt")
sig_genes <- read.table("LUSC_mutsig.sig_genes.txt",header = T,sep="\t")
head(sig_genes,2)
# gene expr reptime hic N_nonsilent N_silent N_noncoding n_nonsilent n_silent n_noncoding nnei x X p q
#1 KEAP1 1996822 207 30 780570 225675 327627 24 0 1 36 117 41857491 0.000000e+00 0.000000e+00
#2 NFE2L2 372300 145 36 770481 203373 353646 28 0 5 16 98 21536829 0.000000e+00 0.000000e+00
#3 PTEN 259678 300 34 535956 124608 917037 16 0 3 45 178 53250450 0.000000e+00 0.000000e+00
#4 CDKN2A 225405 357 -15 422145 109917 240543 26 1 3 6 40 5125035 3.330669e-16 1.570577e-12
#5 TP53 2069567 213 34 546753 150981 948897 148 7 2 1 12 1843632 9.992007e-16 3.769385e-12
#6 MLL2 1770671 214 65 6760515 2174091 3518406 40 7 13 13 72 19828071 5.907712e-09 1.857188e-05
sig_filter<-sig_genes[sig_genes$q<0.05,]$gene
sig_filter
#[1] "KEAP1" "NFE2L2" "PTEN" "CDKN2A" "TP53" "MLL2" "RB1"
#[8] "FBXW7" "PSG2" "HLA-A"
oncoplot(lusc,genes = sig_filter)
整个流程就也算是 SOP,有背景,实操,结果解读,下面大家可以关注,桓峰基因,每日更新,不停息!
References:
1. Lawrence, M. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214-218 (2013).
2. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers [published correction appears in Nature. 2012 Nov 8;491(7423):288.