
点击关注,桓峰基因
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
122篇原创内容
公众号
桓峰基因公众号推出基于基因组变异数据生信分析教程并配有视频在线教程,目前整理出来的教程目录如下:
DNA 1. Germline Mutation Vs. Somatic Mutation 傻傻分不清楚
DNA 2. SCI 文章中基因组变异分析神器之 maftools
DNA 3. SCI 文章中基因组变异分析神器之 maftools
DNA 4. SCI 文章中基因组的突变信号(maftools)
DNA 5. 基因组变异文件VCF格式详解
DNA 6. 基因组变异之绘制精美瀑布图(ComplexHeatmap)
DNA 7. 基因组拷贝数变异分析及可视化 (GISTIC2.0)
DNA 8. 癌症的突变异质性及寻找新的癌症驱动基因(MutSigCV)
DNA 9. 揭秘肿瘤异质性与TMB, MSI之间的相关性
肿瘤异质性指肿瘤内既有致瘤细胞亚群,也有非致瘤细胞亚群。那么肿瘤异质性到底与肿瘤负荷以及微卫星不稳定性到底有什么关系呢?我们这期就用直肠癌的数据进行实际验证一下,方法比较简单,有分析不足的地方,望大家指正!!!
前言
肿瘤异质性是指肿瘤在生长过程中,经过多次分裂增殖,其子细胞呈现出分子生物学或基因方面的改变,从而使肿瘤的生长速度、侵袭能力、对药物的敏感性、预后等各方面产生差异。简而言之,肿瘤异质性指肿瘤内既有致瘤细胞亚群,也有非致瘤细胞亚群。
1. 肿瘤突变负荷 (TMB)
肿瘤突变负荷(tumor mutational burden,TMB)是指在一个特定的肿瘤组织当中相对的基因突变数量,即检测的肿瘤样本中,所评估基因的外显子编码区每兆碱基序列中发生突变的总数。TMB是一个数值,具有高低之分,目前高低TMB的分界值没有统一的标准。
2. 等位基因突变的肿瘤异质性 (MATH)
等位基因突变的肿瘤异质性(mutant-allele tumor heterogeneity,MATH)是基于肿瘤内所有等位基因突变频率(mutant-allele frequencies)来对肿瘤样本的基因异质性进行量化的一种算法,通过计算,每一个样本可以获得一个MATH 值,MATH值的高低反映了肿瘤异质性的高低。
3. 微卫星不稳定性 (MSI)
微卫星不稳定性(Microsatellite Instability,MSI),是指由于在DNA复制时插入或缺失突变引起的微卫星(Microsatellite, MS)序列长度改变的现象。在DNA的复制过程中,DNA聚合酶在遇到串联重复序列时会发生“打滑”,引起微卫星位点中核苷酸的插入或缺失。而这一过程可以被错配修复(mismatch repair,MMR)系统所识别并修复,若MMR基因发生启动子区的高甲基化或编码区的突变,导致其功能丧失,则会无法及时修复微卫星中自发的高频长度变异,从而引起MSI。当DNA错配修复功能(mismreprinter,MMR)异常时,微卫星的复制错误无法纠正并且连续积累,使微卫星的序列长度或碱基组成发生变化,这称为微卫星不稳定性(MSI),并使基因组表现出高突变表型。在肿瘤中,MMR功能缺陷通常是由MMR基因(MLH1,MSH2,MSH6和PMS2)和相关基因EPCAM的致病性突变引起的,或者可能是由MHL1启动子区域的高甲基化引起的缺乏MLH1的表达导致。MS序列可以位于基因的重要非编码区,也可以位于基因的编码区,多态性分布于整个基因组,个体差异大。MSI现象于1993年被Jacobs等人在结直肠癌中首次发现。随着针对MSI的研究深入,发现MSI现象不止存在于结直肠癌,在子宫内膜癌、胃癌、肝细胞癌、乳腺癌等实体瘤中均有发生。
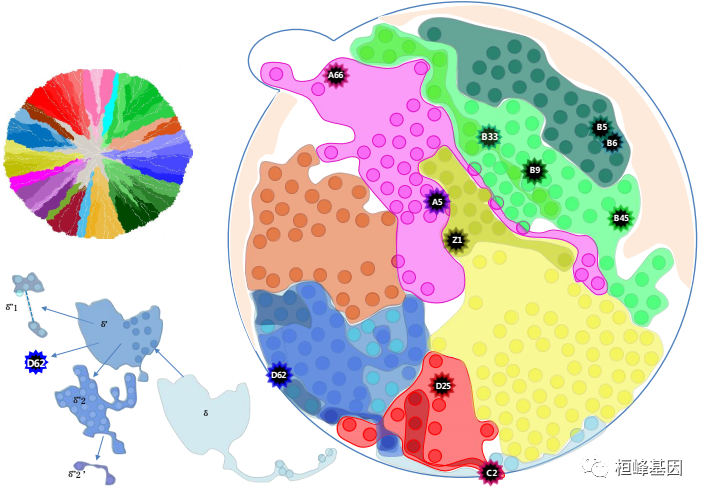
计算方法
1. TMB计算方法
计算公式: TMB(mut/mb)= 总突变数量(包括同义、非同义点突变、置换、插入及缺失突变) / 目标区域编码区大小。
2. MATH计算方法
首先通过测序数据计算每个样本的MAF(mutant-allele fractions)值, 再通过MAF计算得到MAD(median absolute deviation)值,也就是计算每个MAF值与其中位数的绝对差值,并将这些绝对差值的中位数再乘以一个常量(1.4826),从而获得MAD值。最后将MAD值除以MAF的中位数,再乘以100。
3. MSI计算方法
随着高通量测序平台的广泛应用,NGS平台目标区域测序(即NGS panel)或全外显子组测序(WES)或全基因组测序(WGS)开始应用于MSI检测,使用计算工具来同时研究基因组上的大量微卫星序列成为可能,已报道的基于NGS平台的MSI算法包括几种软件,比如:MSIsensor, MANTIS等。
数据下载及整理
1. 软件安装
我们使用TCGAbiolinks软件包下载TCGA-COAD的数据,然后使用maftools对数据进行读取以及计算MATH,TMB等。
if (!require("BiocManager")) install.packages("BiocManager")
if (!require(maftools)) BiocManager::install("maftools")
if (!require(TCGAbiolinks)) BiocManager::install("TCGAbiolinks")
library(TCGAbiolinks)
library(maftools)
2. 数据下载
在计算MATH时,我们需要同时下载突变位点和拷贝数变异数据,以及临床数据来获得MSIscore。
A. 突变位点数据下载
突变数据下载我们只考虑Primary Tumor的样本,选择数据集TCGA-COAD,数据类型等。
querySNV <- GDCquery(project = "TCGA-COAD", data.category = "Simple Nucleotide Variation",
data.type = "Masked Somatic Mutation", workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking",
sample.type = c("Primary Tumor"))
# GDCdownload(querySNV)
B. 拷贝数变异数据下载
CNV数据下载我们同样也只考虑Primary Tumor的样本,选择数据集TCGA-COAD,数据类型等。
queryCNV <- GDCquery(project = "TCGA-COAD", data.category = "Copy Number Variation",
data.type = "Copy Number Segment", sample.type = c("Primary Tumor"))
# GDCdownload(queryCNV)
C. 临床数据下载
临床数据需要我们登录cbioportal下载,当然也可以通过TCGAbiolinks,但是这个方法只能获得MSI的类型,却得不到具体的数值,所以尝试了一下,直接下载更便捷,显著链接如下:
https://www.cbioportal.org/study/summary?id=coadread_tcga_pan_can_atlas_2018
选择charts,点击下载即可下载完成,如下:
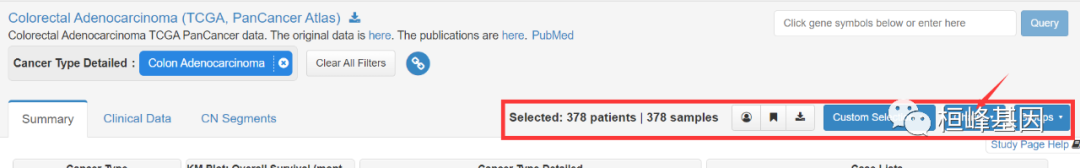
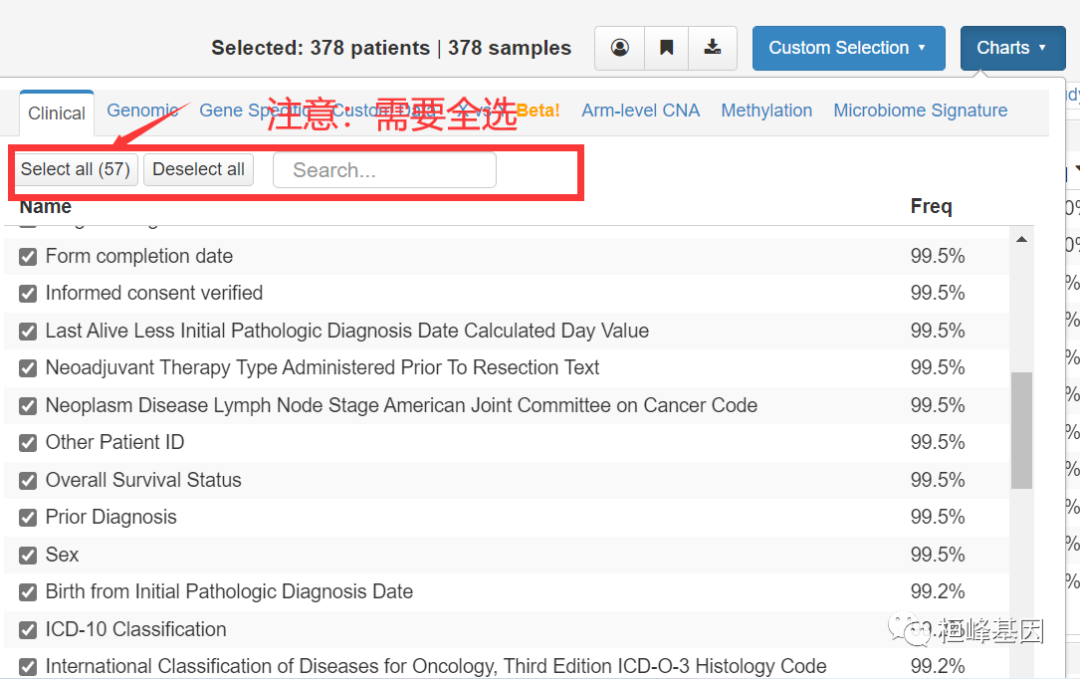
但是下载之后,发现没有MSI数据,于是又重新下载,记得选择Select all,这样才能保证下载到所有的临床信息,如下:
3. 数据整理
下载之后,我们需要对数据进一步整理,目的是将数据中的样本编号保持一致,才可以做后续的分析。
1. 突变数据整理
下载之后通过GDCprepare调取突变表格,并将整理后的数据进行保存,以备后续使用方便!
SNVdata <- GDCprepare(querySNV)
SNVdata$Tumor_Sample_Barcode = substr(SNVdata$Tumor_Sample_Barcode, 1, 12)
SNVdata$t_vaf = SNVdata$t_alt_count/SNVdata$t_depth * 100
SNVdata[1:5, 1:5]
write.table(SNVdata, "coad.maf", row.names = F, sep = "\t", quote = F)
2. CNV数据整理
下载之后通过GDCprepare调取CNV表格,并将整理后的数据进行保存,以备后续使用方便!
CNVdata <- GDCprepare(queryCNV)
head(CNVdata)
CNVdata$Sample = substr(CNVdata$Sample, 1, 12)
seg <- CNVdata[, c(7, 2:6)]
seg = data.table::data.table(seg)
head(seg)
write.table(seg, "coad.seg.txt", row.names = F, sep = "\t", quote = F)
3. 临床数据整理
下载之后整理表格,并将整理后的数据进行保存,以备后续使用方便!
clinData <- read.table("coadread_tcga_pan_can_atlas_2018_clinical_data.tsv", header = T,
sep = "\t")
colnames(clinData)[which(colnames(clinData) == "Patient.ID")] = "Tumor_Sample_Barcode"
4. 数据读取
通过maftools读入突变数据以及临床数据,如下:
coad = read.maf(maf = SNVdata, clinicalData = clinData)
计算TMB, MATH, MSIscore
通过maftools中函数tmb计算TMB,以及绘制TMB分布图。
1. 计算TMB
## 计算TMB
tmb <- tmb(maf = coad, captureSize = 35.8, logScale = T)
head(tmb)
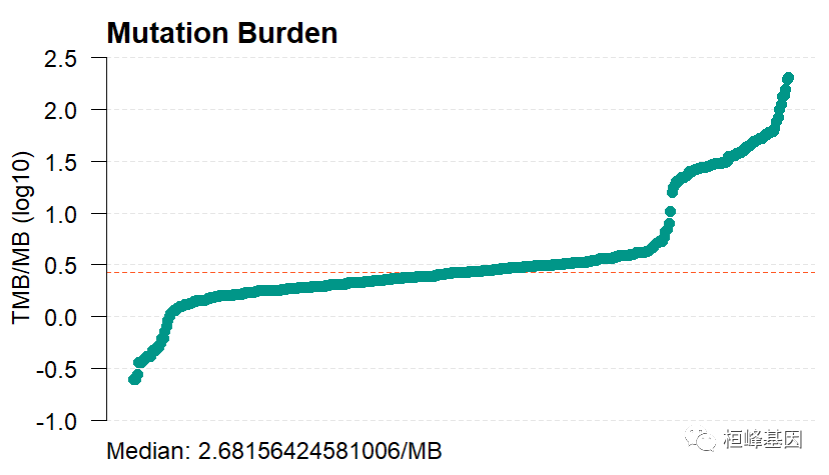
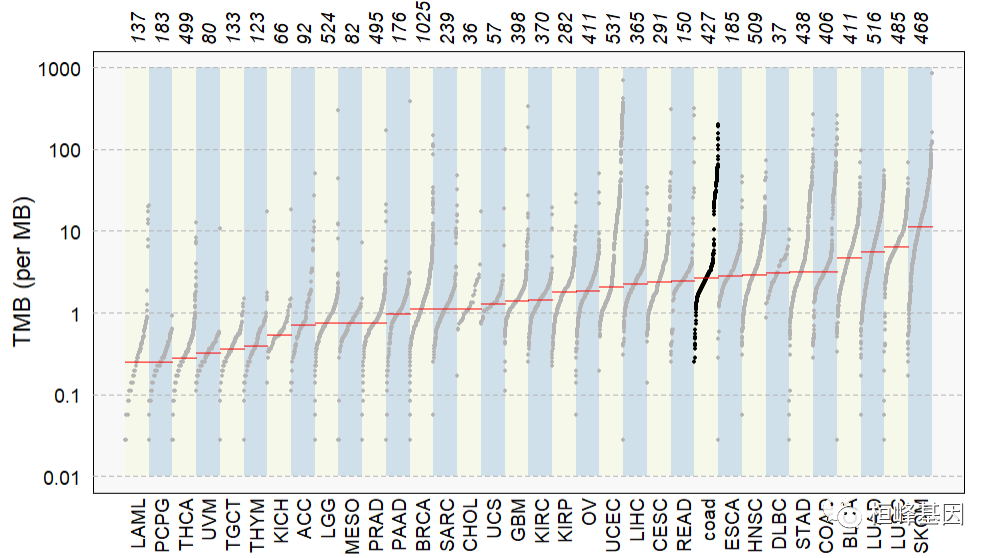
将我们的数据与TCGA泛癌进行比较,我们发现COAD的突变率还是非常高的,由于我们数据只是原发灶的样本,而原始数据中还有复发转移的样本,故我们的结果比数据库所有的样本平均值偏低一些。
laml.mutload = tcgaCompare(maf = coad, cohortName = "coad", logscale = TRUE, capture_size = 35.8)
2. 计算MATH
筛选CNV,SNV以及临床上共有的样本,最后获得419个样本进行后续分析。
samples = unique(getSampleSummary(coad)$Tumor_Sample_Barcode)
seg = seg[Sample %in% samples]
seg_samples = unique(seg[Sample %in% samples]$Sample)
length(seg_samples)
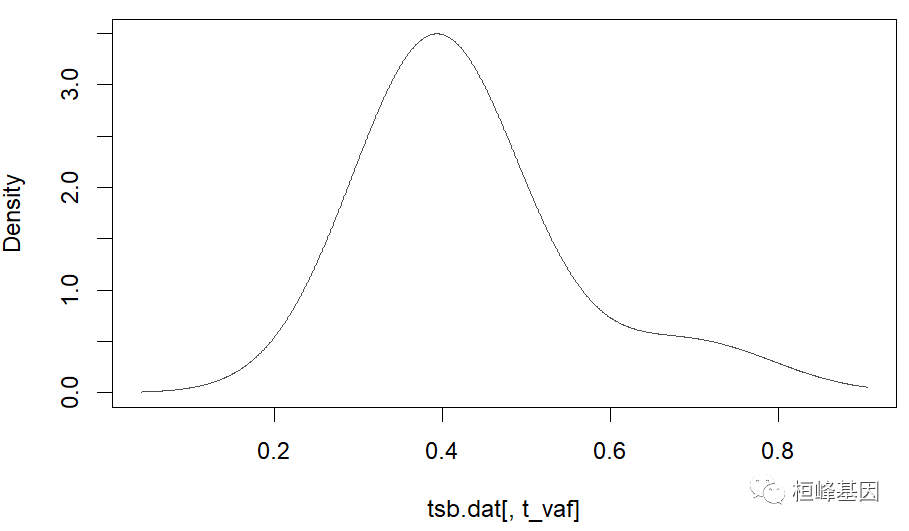
通过maftools软件包中的函数inferHeterogeneity很容易实现MATH的计算以及可视化,我们先来看下单个样本的分析。
library("mclust")
het = inferHeterogeneity(maf = coad, tsb = "TCGA-DM-A28C", vafCol = "t_vaf")
print(het$clusterMeans)
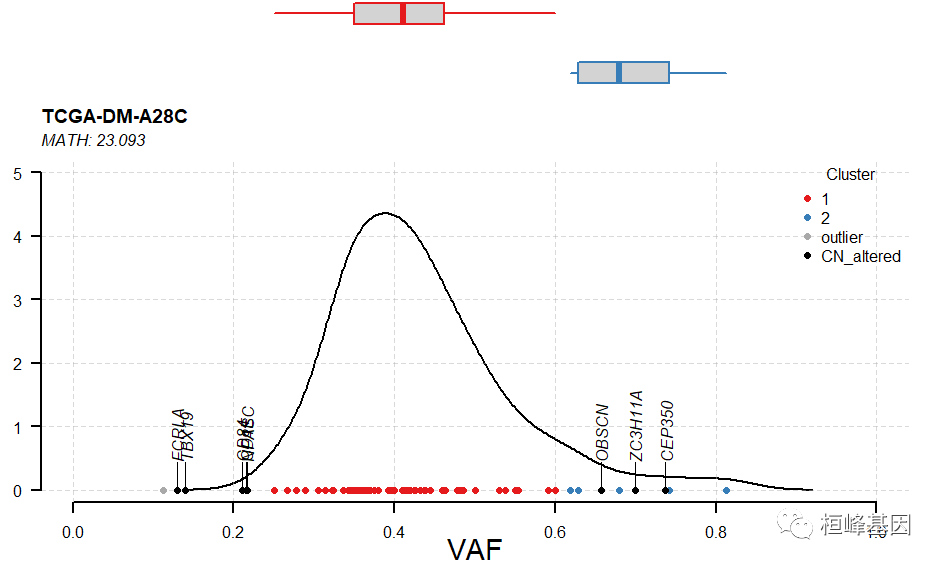
这时我们加入CNV,通过CNV数据的矫正去掉一些落入CNV区域的位点,如下:
segfile = "coad.seg.txt"
het = inferHeterogeneity(maf = coad, tsb = "TCGA-DM-A28C", vafCol = "t_vaf", segFile = segfile)
# Visualizing results. Highlighting those variants on copynumber altered
# variants.
plotClusters(clusters = het, genes = "CN_altered", showCNvars = TRUE)
遍历所有的样本,计算MATH并整理,如下:
outdir = "F:/demo script/基因组分析系列/PlotMATH/"
barcode <- seg_samples
head(barcode)
MATH <- data.frame()
for (i in barcode) {
out.math = inferHeterogeneity(maf = coad, tsb = i, vafCol = "t_vaf", segFile = segfile)
Tumor_Sample_Barcode = unique(out.math$clusterData$Tumor_Sample_Barcode)
m = unique(out.math$clusterData$MATH)
out = data.frame(Tumor_Sample_Barcode, m)
MATH = rbind(MATH, out)
}
head(MATH)
3. 提取 MSIscore
MSI的数据直接在临床数据中获取即可,如下:
MSIdata <- clinData[, c(2, 29, 30, 57)]
head(MSIdata)
4. 合并数据
三组数据都获得后,进行合并,并去掉缺失的样本,如下:
TMB_MATH <- merge(tmb, MATH, by = "Tumor_Sample_Barcode")
clin <- getClinicalData(coad)
tmb_math <- merge(TMB_MATH, MSIdata, by = "Tumor_Sample_Barcode")
tmb_math = na.omit(tmb_math)
head(tmb_math)
4. TMB,MATH,MSIscore相关性
肿瘤异质性与肿瘤负荷,微卫星不稳定之间的关系,通过数值比较的可以更加直观的展现出来,如下:
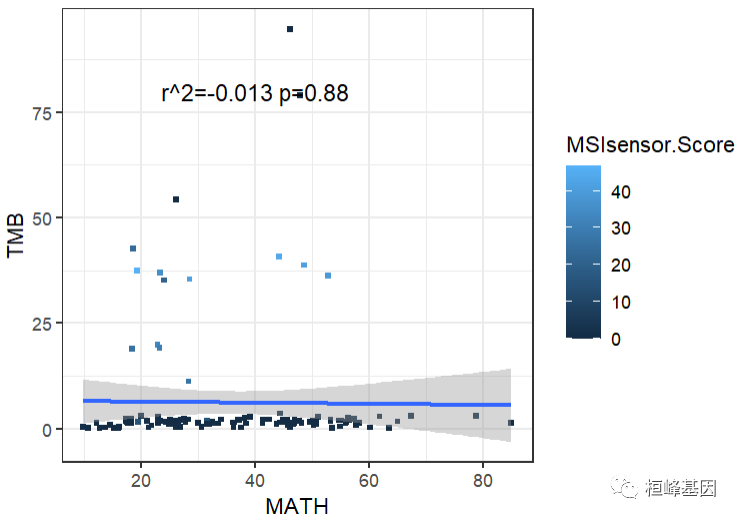
A. TMB与MATH相关性
TMB与MATH略微相关,显著性不高,几乎没有关系。
library(ggplot2)
# TMB与MATH
cor(tmb_math$total_perMB, tmb_math$m, method = "pearson")
cor.test(tmb_math$total_perMB, tmb_math$m, method = "pearson")[3]
p1 <- ggplot(tmb_math, aes(x = m, y = total_perMB)) + geom_point(size = 1, shape = 15,
aes(color = MSIsensor.Score)) + geom_smooth(method = lm) + xlab("MATH") + ylab("TMB") +
annotate("text", x = 40, y = 80, label = "r^2=-0.013 p=0.88", size = 4) + theme_bw() +
guides(fill = guide_colorbar(title = "MSI Score"))
p1
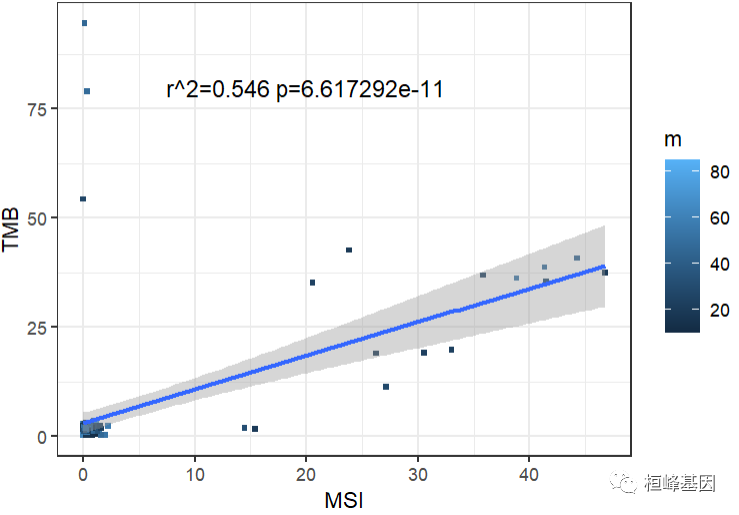
B. TMB与MSI相关性
TMB与MSI正相关,并且非常显著!
cor(tmb_math$total_perMB, tmb_math$MSIsensor.Score, method = "pearson")
cor.test(tmb_math$total_perMB, tmb_math$MSIsensor.Score, method = "pearson")[3]
p2 <- ggplot(tmb_math, aes(x = MSIsensor.Score, y = total_perMB)) + geom_point(size = 1,
shape = 15, aes(color = m)) + geom_smooth(method = lm) + xlab("MSI") + ylab("TMB") +
annotate("text", x = 20, y = 80, label = "r^2=0.546 p=6.617292e-11", size = 4) +
theme_bw() + guides(fill = guide_colorbar(title = "MATH"))
p2
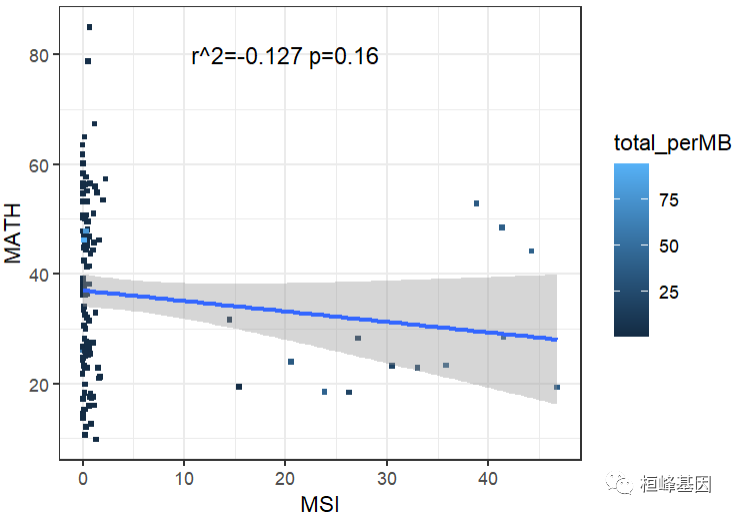
C. MSI与MATH相关性
MSI与MATH略微负相关性,显著性不高,几乎关系不大。
cor(tmb_math$m, tmb_math$MSIsensor.Score, method = "pearson")
cor.test(tmb_math$m, tmb_math$MSIsensor.Score, method = "pearson")[3]
p3 <- ggplot(tmb_math, aes(x = MSIsensor.Score, y = m)) + geom_point(size = 1, shape = 15,
aes(color = total_perMB)) + geom_smooth(method = lm) + xlab("MSI") + ylab("MATH") +
annotate("text", x = 20, y = 80, label = "r^2=-0.127 p=0.16", size = 4) + theme_bw() +
guides(fill = guide_colorbar(title = "TMB"))
p3
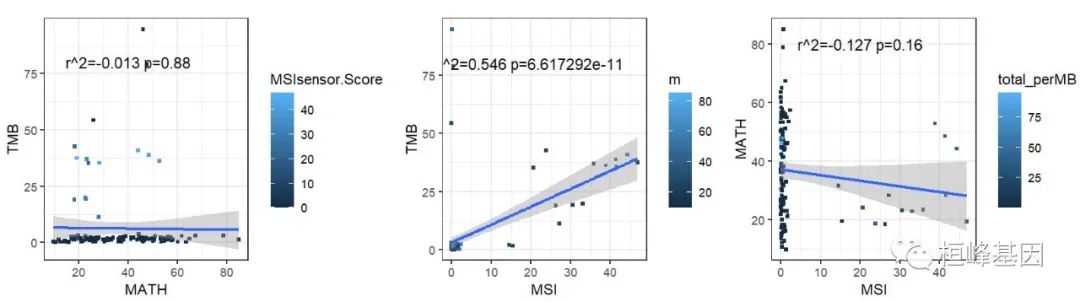
将三个结果组合一起,仔细观察:
library(patchwork)
p1 | p2 | p3
结果解读
从相关性结果上来看,TMB与MSI关系非常密切,而TMB与MATH异质性相关性不高,MSI同样与MATH相关性不高,但是这三个变量是否能作为COAD癌症的预后标志物,请听下次分解!
References:
-
Ling S, Hu Z, Yang Z, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution [published correction appears in Proc Natl Acad Sci U S A. 2016 Feb 2;113(5):E663]. Proc Natl Acad Sci U S A. 2015;112(47):E6496-E6505. doi:10.1073/pnas.1519556112
-
Mroz EA, Rocco JW. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol. 2013;49(3):211-215. doi:10.1016/j.oraloncology.2012.09.007