使用PAML进行分歧时间计算
PAML软件中的mcmctree命令可以使用Bayesian方法估算物种分歧时间。
对程序输入带有校准点信息的有根树、多序列比对结果,即可得到进化树各内部节点95%置信区间分歧时间信息。
PAML软件也能利用带有枝长信息的树,快速进行分歧时间计算。
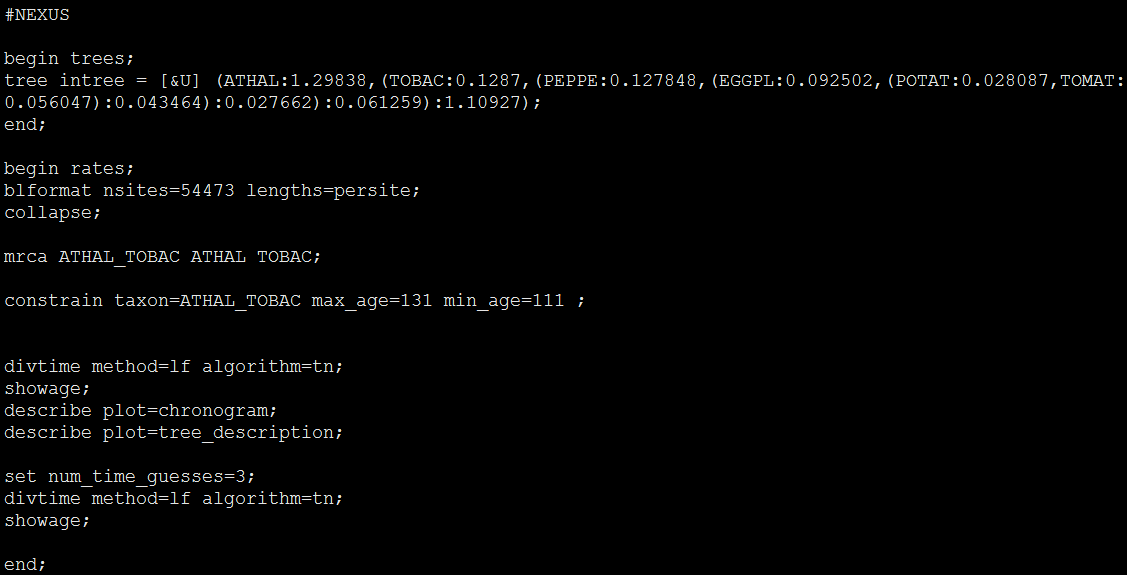
1. 输入文件(1/3):带有校准点的有根树 input.trees 输入一个带有校准点的有根树文件,示例如下:
7 1
((((human, (chimpanzee, bonobo)) '>.06<.08', gorilla), (orangutan, sumatran)) '>.12<.16', gibbon);
文件内容分两行:第一行表述树中有7个物种,共计1个树,两个数值之间用空分割;
第二行则是Newick格式树信息,其中包含有校准点信息。校准点信息一般指95%HPD(Highest Posterior Density)对应的置信区间;
校准点单位是100MYA(软件说明文档中使用该单位,也推荐使用该单位,若使用其它单位,后续配置文件中的相关参数也需要对应修改)。此外,Newick格式的树尾部一定要有分号,没有的话程序可能不能正常运行。
2. 输入文件(2/3):密码子在3个位点的多序列比对结果(Phylip格式) input.txt
7 3331 human AGACTGTTAGCAGCGCCGGGCAACTCCCTACTCAA... chimpanzee AGACTGTTGGCAACGTCGGGCAACTCCCCGCCCAA... bonobo AGACTGTTGGCAACGCCGGGCAACTCCCCGCCCAA... gorilla AGATTGTTAGCAACGTCGGGTAACCCCCCACTCAA... orangutan AGGCTACTAACAGCGCCGGACGATTCCTCGCCTAA... sumatran AGACTACTAACAGCGCCGGGCGATTCCTCACCCAA... gibbon AGACTATTGACAACGTCGGGCAACTCTCTACTCAA... 7 3331 human AAATTCCTTCCCTTGTCCCTTTTTTCCTTTCATTA... chimpanzee AAATTCCTCCCCTTGTCCCTTTTTTCCTTTCATTA... bonobo AAATTCCTCCCCTTGTCCCTTTTTTCCTTTCATTA... gorilla AAATTCCTTCCCTTGTCCCTTTTTTCCTTTCATTA... orangutan AAATTCCTCCCCTTGTCCCTTTTTTCCTTTCATTA... sumatran AAGTTCCTTCCCTTGTCCCTTTTTTCCTTTCATTA... gibbon AAATTCCTCCCCTTGTCCCTCTTTTCCTTTCATTA... 7 3331 human CATGCTACTCCACACACCAAGCTATCTAGCCTCCC... chimpanzee CATACTACTCCACACACCAAACTACCTAGCCTCCC... bonobo CATGCTACTCCACACACCAAGCTACCTAGTCTCCC... gorilla CATACTACTCCACACACCAAATCATCTAGCCTCCC... orangutan CATACCACTCCACACCCTATACCATCCAACTTCCC... sumatran CATATCACTCCAAACCCCAAACCATCCAGCCTCCC... gibbon CATACTACTCCATACACCAAATTATCCAACTCCCC...
PAML要求输入的Phylip格式,其物种名和后面的序列之间至少间隔两个空格(是为了允许物种名的属名和种名之间有一个空格)。
当然,也可以输入一个多序列比对结果进行分析。在软件说明文档中,是密码子三个位点分别的多序列比对结果,程序可以分别计算密码子各个位点的变异速率。
此外,也可以输入密码子或氨基酸序列比对信息,则需要再配置文件中修改相应的参数来表明数据类型。若使用氨基酸序列来进行分析,由于mcmctree命令不能选择较好的氨基酸替换模型进行分析,需要自己手动运行codeml进行分析后,在生成中间文件用于运行mcmctree,比较麻烦。因此,推荐按上述方法使用密码子三位点的碱基序列作为输入信息进行PAML分析。
3. 输入文件(3/3):mcmctree命令的配置文件 mcmctree.ctl
软件难点在于配置文件的理解,示例如下:
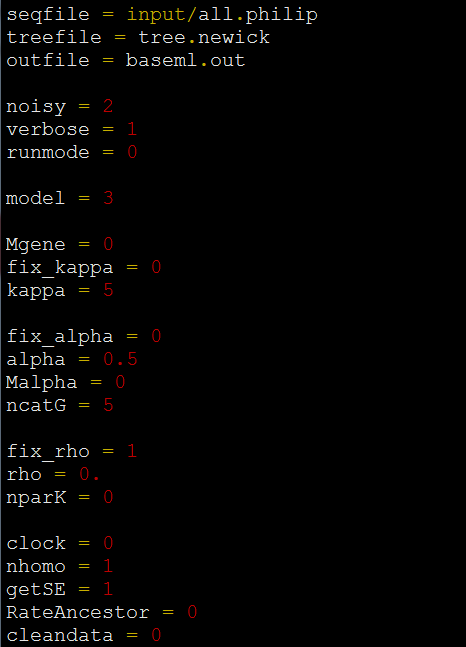
*输入输出参数:
seed = -1 *设置一个随机数来运行程序。若设置为-1,表示使用系统当前时间为随机数。
seqfile = input.txt *设置输入的多序列比对文件路径
treefile = input.trees *设置输入的带校准点信息有根树的文件路径
mcmcfile = mcmc.txt *设置输出的mcmc信息文本文件,可以用Tracer软件查看
outfile = out.txt *设置输出文件路径,该文件中记录了超度量树和进化速率等参数信息。
*数据使用说明参数:
ndata = 3 *设置输入的多序列比对的数据个数。这里指密码子3个位置的数据。
seqtype = 0 *设置多序列比对的数据类型:
0,表示核酸数据;
1,表示密码子比对数据;
2,表示氨基酸数据。
根据不同的数据类型,程序调用相应的baseml或codeml进行相应的参数计算。
usedata = 1 *设置是否利用多序列比对的数据:0,表示不使用多序列比对数据,则不会进行likelihood计算,虽然能得到mcmc树且计算速度飞快,但是其分歧时间结果是有问题的;
1,表示使用多序列比对数据进行likelihood计算,正常进行MCMC,是一般使用的参数;
2,进行正常的approximation likelihood分析,此时不需要读取多序列比对数据,直接读取当前目录中的in.BV文件。该文件是使用usedata = 3参数生成的out.BV文件重命名而来的。
此外,由于程序BUG,当设置usedata = 2时,一定要在改行参数后加 *,否则程序报错 Error: file name empty..
3,程序利用多序列比对数据调用baseml/codeml命令对数据进行分析,生成out.BV文件。
由于mcmctree调用baseml/codeml进行计算的参数设置可能不太好(特别时对蛋白序列进行计算时),推荐自己修该软件自动生成的baseml/codeml配置文件,
然后再手动运行baseml/codeml命令,再整合其结果文件为out.BV文件。
cleandata = 0 *设置是否移除不明确字符(N、?、W、R和Y等)或含以后gap的列后再进行数据分析:
0,不需要,但在序列两两比较的时候,还是会去除后进行比较;
1,需要。
clock = 2 *设置分子钟方法:
1,global clock方法,表示所有分支进化速率一致;
2,independent rates方法,各分支的进化速率独立且进化速率的对数log(r)符合正态分布;
3,correlated rates方法,和方法2类似,但是log(r)的方差和时间t相关。
* TipDate = 1 100 *当外部节点由取样时间时使用该参数进行设置,同时该参数也设置了时间单位。具体数据示例请见examples/TipData文件夹。
RootAge = '<1.0' *设置root节点的分歧时间,一般设置一个最大值。
*位点替换模型参数:
model = 4 *设置碱基替换模型:0,JC69;1,K80;2,F81;3,F84;4,HKY85。
当设置usedata = 1时,不能使用其它碱基替换模型。
若需要选择其它碱基替换模型,则考虑使用usedata = 3参数,就可以设置model参数值为其它的碱基替换模型。
此时,程序会将数据进行分割,例如,ndata = 3,则程序分别对3个数据进行分析,生成baseml的输入文件,
并调用baseml进行分析(若数据量较大,分析较慢,推荐按ctrl + c 强行终止,程序则终止baseml的运行,进行下一个数据的输入文件生成并运行下个数据的baseml命令,
再按 ctrl + c 强行终止,这样可以让程序生成3各数据的输入文件和baseml配置文件,然后手动并行化运行,加快时间);
每个分析结果中会得到rst2结果文件;可以在此处手动修改tmp0001.ctl配置文件,例如,修改其model参数值,推荐修改outfile参数值,从而可以手动并行化运行baseml命令;
然后将所有数据的rst2结果使用cat命令合并成文件in.BV,用于下一步设置参数usedata = 2,进行MCMC分析。
alpha = 0.5 *核酸序列中不同位点,其进化速率不一致,其变异速率服从GAMMA分布。一般设置GAMMA分布的alpha值为0.5。
若该参数值设置为0,则表示所有位点的进化速率一致。
伽玛分布(Gamma Distribution)是统计学的一种连续概率函数,是概率统计中一种非常重要的分布。“指数分布”和“χ2分布”都是伽马分布的特例。 Gamma分布中的参数α称为形状参数(shape parameter)
Gamma分布的特殊形式,当形状参数α=1时,伽马分布就是参数为γ的指数分布,X~Exp(γ)当α=n/2,β=1/2时,伽马分布就是自由度为n的卡方分布,X^2(n)
此外,当userdata = 2时,alpha、ncatG、alpha_gamma、kappa_gamma这4个GAMMA参数无效。
因为userdata = 2时,不会利用到多序列比对的数据。
ncatG = 5 *设置离散型GAMMA分布的categories值。
BDparas = 1 1 0.1 *设置出生率、死亡率和取样比例。若输入有根树文件中的时间单位发生改变,则需要相应修改出生率和死亡率的值。
例如,时间单位由100Myr变换为1Myr,则要设置成".01 .01 0.1"。
kappa_gamma = 6 2 *设置kappa(转换/颠换比率)的GAMMA分布参数。
alpha_gamma = 1 1 *设置GAMMA形状参数alpha的GAMMA分布参数。
*进化速率参数:
rgene_gamma = 2 20 1 *设置序列中所所有位点平均[碱基/密码子/氨基酸]替换率的Dirichlet-GAMMA分布参数:
alpha=2、beta=20、初始平均替换率为每100million年(取决于输入有根树文件中的时间单位)1个替换。
若时间单位由100Myr变换为1Myr,则要设置成"2 2000 1"。
总体上的平均进化速率为:2 / 20 = 0.1 个替换 / 每100Myr,即每个位点每年的替换数为 1e-9。
sigma2_gamma = 1 10 1 *设置所有位点进化速率取对数后方差(sigma的平方)的Dirichlet-GAMMA分布参数:alpha=1、beta=10、初始方差值为1。
当clock参数值为1时,表示使用全局的进化速率,各分枝的进化速率没有差异,即方差为0,该参数无效;
当clock参数值为2时,若修改了时间单位,该参数值不需要改变;
当clock参数值为3时,若修改了时间单位,该参数值需要改变。
finetune = 1: .1 .1 .1 .1 .1 .1 *冒号前的值设置是否自动进行finetune,一般设置成1,然后程序自动进行优化分析;
冒号后面设置各个参数的步进值:times, musigma2, rates, mixing, paras, FossilErr。
由于有了自动设置,该参数不像以前版本那么重要了,可能以后会取消该参数。
*MCMC参数:
print = 1 *设置打印mcmc的取样信息:
0,不打印mcmc结果;
1,打印除了分支进化速率的其它信息(各内部节点分歧时间、平均进化速率、sigma2值);
2,打印所有信息。
burnin = 2000 *将前2000次迭代burnin后,再进行取样(即打印出该次迭代计算的结果信息,各内部节点分歧时间、平均进化速率、sigma2值和各分支进化速率等)。
sampfreq = 10 *每10次迭代则取样一次。
nsample = 20000 *当取样次数达到该次数时,则取样结束(程序也将运行结束)。
运行mcmctree命令进行分歧时间计算
程序运行命令:
mcmctree mcmctree.ctl
结果文件解释
程序在运行过程中,会在屏幕生生成一些信息。
比较耗时间的步骤主要在于取样的百分比进度:
第一列:取样的百分比进度。
第2~6列:参数的接受比例。一般,其值应该在30%左右。20~40%是很好的结果,15~70%是可以接受的范围。若这些值在开始时变动较大,则表示burnin数设置太小。
第7~x列:各内部节点的平均分歧时间,第7列则是root节点的平均分歧时间。若有y个物种,则总共有y-1个内部节点,从第7列开始后的y-1列对应这些内部节点。
倒数第3~x列:r_left值。若输入3各多序列比对结果,则有3列。x列的前一列是中划线 - 。
倒数第1~2列:likelihood值和时间消耗。
屏幕信息最后,给出各个内部节点的分歧时间(t)、平均变异速率(mu)、变异速率方差(sigma2)和r_left的Posterior信息:
均值(mean)、95%双侧置信区间(95% Equal-tail CI)和95% HPD置信区间(95% HPD CI)等信息。
此外,倒数第二列给出了各个内部节点的Posterior mean time信息,可以用于收敛分析。
在当前目录下,生成几个主要结果:
FigTree.tre 生成含有分歧时间的超度量树文件
mcmc.txt MCMC取样信息,包含各内部节点分歧时间、平均进化速率、sigma2值等信息,可以在Tracer软件中打开。
通过查看各参数的ESS值,若ESS值大于200,则从一定程度上表示MCMC过程能收敛,结果可靠。
out.txt 包含由较多信息的结果文件。例如,各碱基频率、节点命名信息、各节点分歧时间、进化速率和进化树等。
使用approximate likelihood方法更快速地计算分歧时间
按照以上方法进行MCMC计算非常消耗计算。
为了加快计算速度,可以将分析分成两个步骤:
(1)首先,使用Maximum Likelihood方法计算枝长和Hessian信息;
(2)再使用MCMC方法计算分歧时间。
运行步骤也相应分成两步:
(1)设置配置文件中参数 usedata = 3,然后和上面同样运行mcmctree命令,软件会对各个多序列比对结果调用baseml/codeml命令进行分歧时间计算,生成结果文件rst2,将各个多序列比对结果文件内容直接cat(linux系统的常用命令)合并成out.BV文件;
(2)然后将out.BV文件重命名为in.BV,并将配置文件中参数 usedata = 2,再次运行同样的mcmctree命令,程序会运行MCMC分析并得到结果。使用该方法,其MCMC速度快很多。
若输入数据是protein序列,则mcmctree调用codeml命令进行分析时的model参数选择是不正确的,需要自己手动修改codeml命令的参数配置文件后再运行codeml命令,并将其结果文件rst2的内容合并到out.BV文件中。
若想使用更复杂的模型,如GTR进行mcmctree分析,则按同样方法手动修改配置文件并允许baseml命令,最后将rst2的内容整合到out.BV文件中。此外,可以将分别来自核酸(非编码RNA)和蛋白的rst2内容同时整合到out.BV中进行计算。
使用infinitesites输入含有枝长信息的系统进化树快速进行分歧时间计算
infinitesites程序进行分歧时间计算时,其假设前提为多序列比对的序列长度为无限长。
相比于正常的mcmctree命令,要求额外多输入一个名为 FixedDsClock23.txt 的文件。该文件中存放了相应的带有枝长信息的系统发育树。该文件中系统发育树的拓扑结构要和input.trees一致。推荐使用baseml命令输入input.trees和多序列比对结果计算枝长。
FixedDsClock23.txt 文件内容示例:
7 ((((human: 0.029043, (chimpanzee: 0.014557, bonobo: 0.010908): 0.016729): 0.015344, gorilla: 0.033888): 0.033816, (orangutan: 0.026872, sumatran: 0.022437): 0.069648): 0.073309, gibbon: 0.024637); ((((human: 0.012463, (chimpanzee: 0.002782, bonobo: 0.003835): 0.003331): 0.004490, gorilla: 0.014278): 0.006308, (orangutan: 0.010818, sumatran: 0.008845): 0.030551): 0.004363, gibbon: 0.029246); ((((human: 0.270862, (chimpanzee: 0.066698, bonobo: 0.056883): 0.124104): 0.139082, gorilla: 0.310797): 0.391342, (orangutan: 0.152555, sumatran: 0.114176): 0.696518): 0.017607, gibbon: 1.394718);
运行infinitesites命令进行分歧时间计算:
infinitesites mcmctree.ctl使用infinitesites进行分歧时间计算时,程序要求输入多序列比对文件。虽然程序读取了序列信息,但在计算时会忽略其序列信息。
其它注意事项
(1) 如何检测MCMC的结果是否达到收敛状态?
收敛的意思,即经过很多次迭代后,得到的MCMC树的各枝长趋于一个定值,变动非常小。
于是,最直接的检测方法是:分别使用不同的Seed值进行mcmctree或infinitesites进行两次或多次分析,然后比较两个结果树是否一致,实际就是比较树文件中各内部节点的Height值(分歧时间 / Posterior time)。
比较方法可以有多种:
(a) 结果文件mcmc.txt中每行记录了一个取样的结果,包含各内部节点的Posterior time值,即进化树的Height值,对该mcmc.txt文件进行分析,得到每个内部节点的Posterior mean time值。然后作图 (官方说明文档第6页图示),横坐标表示第一次运行的各内部节点的分歧时间均值、纵坐标表示第二次运行的各内部节点的分歧时间均值。该散点图要表现出非常明显的对角线,才认为达到收敛。这时可以考虑计算其相关系数来判断是否符合线性。
(b) 我觉得第一种官方文档给的方法比较麻烦且是否符合对角线没有明确的定义,于是就直接比较两次结果树文件中的各枝长。计算各枝长总的偏差百分比,当偏差百分比低于0.1%,则认为两次结果非常吻合,差异低于0.1%,认为达到收敛。
(c) 此外,还可以使用Tracer分析mcmc.txt文件,检测其ESS值,一般认为该值高于200,则可能达到收敛。该方法可用于辅助检测。最后,若不收敛,则需要提高burnin、nsample值,重新运行程序。
(2) 如何设置burnin、sampfreq和nsample值?
(a) 一般推荐设置nsample值不小于20k(官方示例数据中设置的值),只有当该值较大时,求得的均值才有意义。
(b) sampfreq表示取样间隔,一般设置为10,100或1000。nsample 和sampfreq 的乘积表示有效迭代的次数,这些迭代越准确,次数越大,则结果越好,越趋于收敛。当然,次数越大,越消耗时间。若数据量很小,可以考虑设置sampfreq为1000;若数据量大,每次迭代耗时多,则考虑设置sampfre为10。
(c) 一般最开始的迭代结果很差,需要去除(burnin)其结果,则设置burnin值。要设置足够大的burnin值,保证在程序运行时当迭代比例达到0% (即burnin结束) 时,其参数的接受比例值较好,在30%左右且随迭代次数增多时变动幅度不大。推荐设置burnin的迭代次数为有效次数的10~40%。PAML软件时先burnin后再记录有效迭代的参数值;其它软件如BEAST则记录所有的参数值后,最后汇总时burnin掉一定比例的数据。
(d) 总体上,其实就是两个参数:burnin掉的迭代次数和用于计算结果的有效迭代次数。由于需要根据有效迭代数据来进行均值计算,若记录每次迭代的参数,则生成的mcmc.txt文件行数过多,文件太大,汇总时也消耗计算。于是设置没隔一定迭代轮数取样一次,这样生成的mcmc.txt文件不会太大,对最后的均值计算影响不大。
(e) 我个人推荐有效迭代次数 1~10M 次,burnin 0.2~4M次。按时间消耗从少到多的参数值:
数据简单时,进行0.5M迭代次数,burnin比例40%:{ burnin 200k; sampfreq 10; nsample 50k }
数据中等时,进行1M迭代次数,burnin比例40%:{ burnin 400k; sampfreq 10; nsample 100k }
数据复杂时,进行5M迭代次数,burnin比例20%:{ burnin 1000k; sampfreq 10; nsample 500k }
数据巨大时,进行10M迭代次数,burnin比例20%:{ burnin 2000k; sampfreq 100; nsample 100k }
使用相应参数进行分析后,若不满意其收敛程度,则选用更多迭代次数的参数。
从DNA序列到分化时间——如何构建带分歧时间的进化树
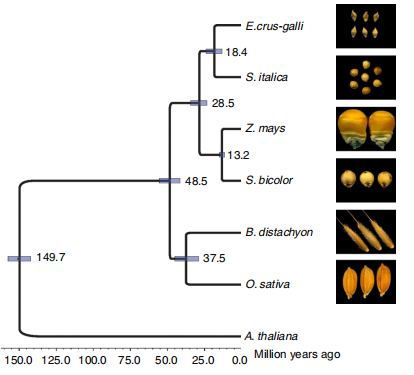
分支带有物种的分化时间,有些还可添加分化时间的置信度范围
根据分子钟理论,基因序列中密码子随着时间的推移以几乎恒定的比例相互替换,即具有恒定的演化速率,两个物种之间的遗传距离将与物种的分化时间成正比。我们通常采用单拷贝基因家族中的四重简并位点来估算分子钟(替换速率)以及物种间的分化时间,密码子中的四重简并位点由于第三碱基不改变所编码的氨基酸,属于中性进化,其中中性替换速率一般用每个位点每年的变异数来衡量。
利用贝叶斯统计或其他方法估计物种分歧时间的程序很多,如R8S、MCMCTREE、MULTIDIVTIME、BEAST等,通过不同的策略将化石时间信息整合到一个系统发育树中,从而计算得到Divergence time Tree。
下面我们对构建Divergence time Tree的方法和步骤做一个简述:
利用单拷贝基因的CDS或PEP序列构建系统发育树,推荐PhyML或Mrbayes 。因为ML树和贝叶斯进化树对核苷酸 / 氨基酸替代模型的选择非常敏感,故在进行进化树或分化时间构建之前,需对核苷酸 / 氨基酸替代模型进行选择
构建获得 newick格式的进化树,如下例子:
![]()
02获得fossil calibration time
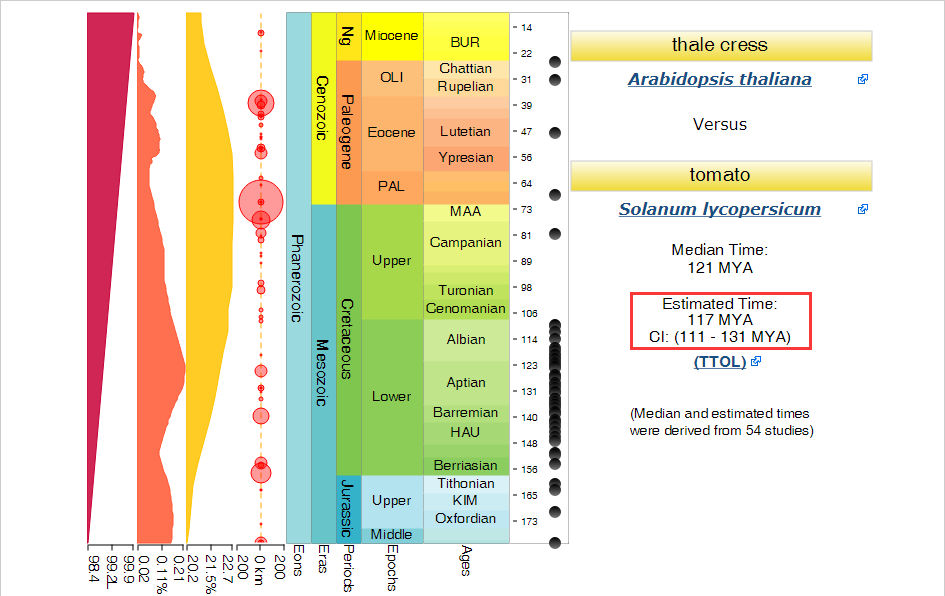
那如何查两两物种间的化石分化时间?
通过网站http://www.timetree.org/ ,该网站根据多篇文献支持提供两两物种间的分化时间,并给出置信度范围,单位是Mya(million years ago)。
另外一个可以查询分化时间的网站是https://fossilcalibrations.org/ ,可以互相参考。
在已经构建好的树中添加化石时间,主要形式是 '>0.22<6.75','>'后接分化时间的最小值,'<'后接最大值,如下实例:
![]()
当样本比较多时,要考虑不同距离的节点,只有一个节点估算的可能不准确,可以提供三到五个的节点(物种)的分化时间。
03分歧时间估计
这里主要介绍3种程序:
1. 使用 r8s(https://sourceforge.net/projects/r8s/)估计分歧时间,速度很快,一般时间<10min
准备配置文件r8s.ctl:
运行程序:
![]()
2. 使用multidivtime(https://brcwebportal.cos.ncsu.edu/thorne/multidivtime.html) 估计分歧时间,时间~5hours
-> 先利用PAML baseml估计分支的核苷酸频率、转换/颠换率等参数
再利用paml2modelinf将baseml输出文件转换为estbranches输入文件
![]()
利用estbranche对树分支长度进行最大似然率估计
![]()
输入文件是前一步的输出结果
![]()
![]()
-> 运行multidivtime进行分歧时间估计
利用前一步的输出,准备配置文件divtime.ctl,运行程序
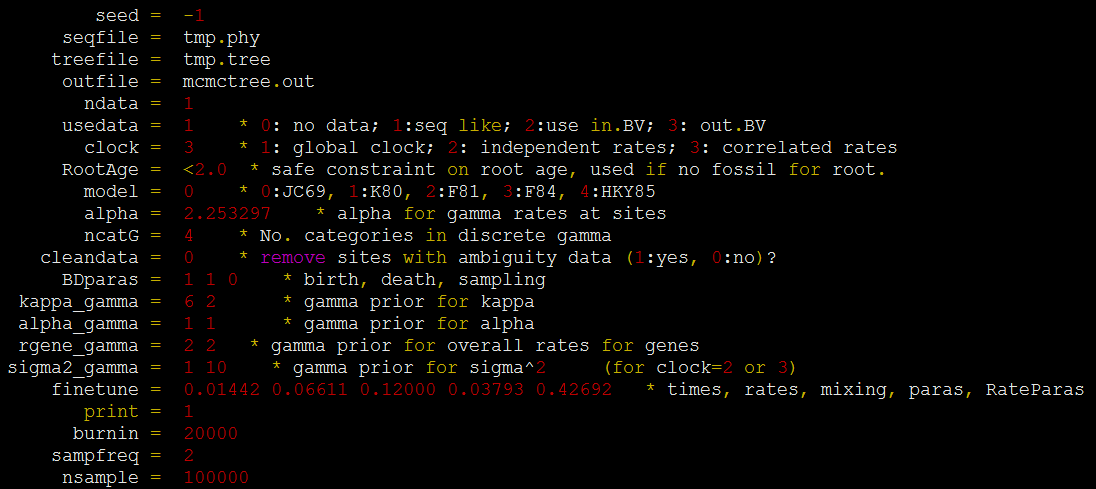
3. 使用 PAML mcmctree(http://abacus.gene.ucl.ac.uk/software/paml.html) 估计分歧时间,强烈推荐,时间~20hours
准备配置文件 mcmctree.ctl
![]()
运行程序:
计算完成后,就获得了带分歧时间的进化树了,后续可以对进化树进行一系列美化操作。
来源
http://www.chenlianfu.com/?p=2974
https://www.biomart.cn/65423/news/2908793.htm