原文链接 https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201703114
1.简介
近年来,开发更有效但同样可靠的原子势以达到对复杂系统的原子级理解已经受到相当大的关注。一个有希望的新发展是引入机器学习(ML)方法来描述原子相互作用。本综述考虑了采用人工神经网络的一类重要的ML电位的方法论。
新分子和新材料的设计必须基于对它们的性质与原子性以及电子结构之间关系的基本理解,它们的动力学特性和它们的反应性,这些都与原子和分子的相互作用密切相关。
随着技术发展,获取的实验数据越来越庞大,然而,如果没有计算机模拟的补充信息,就不可能解释这些丰富的实验数据,如果没有实验和理论的共同努力,就不可能在原子水平上有全面的理解。因此,开发适用于日益复杂的系统的理论工具尤为重要,其最终目标是实现预测能力。
DFT在含约1000个原子的凝聚体系的理论研究中占据了核心地位。另一方面,对于中等大小的分子,可以使用一种非常精确的基于波函数的方法层次结构,这种方法具有良好的误差控制能力,通常可以与最佳实验数据相媲美。DFT和波函数方法在帮助理解化学、物理和材料科学中的许多问题方面都取得了巨大的成功。
为了跟上当前研究的需求,有必要放弃理想化的模型系统,搜寻范围更广的材料、结构和成分,跟踪系统在相当长的时间尺度上的演化,高精度地解决原子相互作用的细微细节,如反应障碍。这种对更大系统和更长的模拟的需求并不新鲜,而且已经挑战了几代理论家。
这些问题包括材料科学,水溶液中涉及水分子离解和重组的反应,以及界面上的化学过程。所有这些系统都要求可靠地描述键断裂和键生成,以及复杂的键合情况、原子环境和结构重排。在第一原理计算中很自然地考虑了这一切,但在另一方面,这些应用仍然缺乏有效的原子势。因此,为了避免对这类系统进行电子结构计算,需要改进原子势,使原子位置与势能之间具有直接的函数关系,即势能面(PES),而势能面通常是一个复杂的高维函数。量子力学解决这一问题,但通常给出PES的解析形式并不容易。最重要的要求:

- 一种非常高的精度,可与第一性原理方法相媲美,包括高阶多体效应
- 描述化学反应和任意原子构型的能力
- 一个非常高效的模拟大型系统
- 适用于所有类型的键合和原子相互作用,从通过共价键的色散相互作用到金属键合
- 拥有系统改进、验证和错误控制的策略
- 一个通用的自动构造协议
- 没有特别的近似或系统特有的能量贡献,这限制了对某些类型系统的适用性
(通过引入物理近似来获取降低计算成本)尽管这些“物理势”有许多成功的应用,但近年来原子间势的构造发生了根本性的变化。在新一代势函数中,不再使用物理近似,而是采用非常灵活的函数形式来拟合高级电子结构数据。如今,机器学习(ML)方法在这门“数学势能”课程中处于领先地位。
在这篇综述中,我们将用神经网络电位(NNPs)来说明开发准确有效的ML电位所面临的挑战和解决方案。
2.机器学习势能位
ML方法在化学和物理领域也长期成功应用,主要用于数据处理和分类,如NMR和质谱分析、结晶动力学建模、和蛋白质结构预测等。 优势:几个数量级的准确性&一定的通用性普适性。
ML potentials (MLPs) 定义如下三个条件:1.在MLPs中,结构-能量关系用ML方法表示 2.MLPs是由一组相一致的第一性原理能量(或 力)而构造的。 3.除了用于生成参考数据集的电子结构方法外,MLPs不包含任何物理近似或特别假设。
由于MLPs能够在原子位置和能量之间建立直接的关系,因此它可以处理任意的原子构型,而不需要指定键或原子类型,这是简单力场的一个主要限制。这种普适性使得mlp在传统潜力极具挑战性的系统中具有广阔的应用前景。
构造MLP的步骤:
- 首先,选择一种电子结构方法作为参考,它能够捕获系统的正确物理行为。
- 第二步,为训练ML方法准备数据。这是通过将原子位置转换为一组特殊的坐标来实现的。
- 然后,拟合过程,调整ML方法的参数,尽可能准确地再现参考数据。
- 一旦仔细检查过,就可以应用了。

3.神经网络势能位
人工智能/神经网络的起起落落......
多年来,许多不同类型的NNs被开发出来并用于不同的目的。除了对数据分类具有良好的适用性外,已经证明NNs在原理上能够以任意精度表示未知的多维实值函数。这一性质是其适用于势能面构建的两个理论基础之一。第二个是量子力学的玻恩奥本海默近似,它指出一个系统的能量可以作为核位置的参数函数给出。
3.1前馈神经网络的早期电位
所有的神经网络电位都包含一个或多个前馈神经网络。这些用来定义系统结构和期望性质之间的关系,在PES的情况下,期望性质是系统的势能。

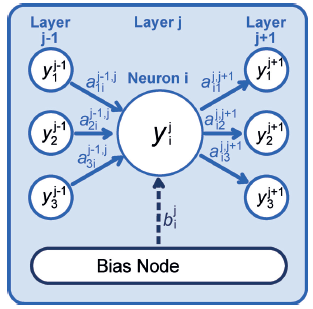
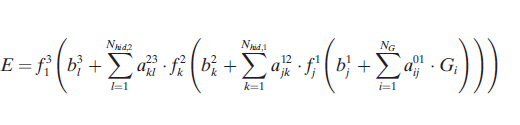
除了由{Gi}指定的原子位置外,系统的能量依赖于连接权值参数{a}和偏置权值{b}的数值。这些参数是在基于梯度的迭代优化中确定的,使用一组已知的参考能量—有时是力—来自电子结构计算。这种优化的总体目标是最小化给定训练集的能量(和力)的均方根误差(RMSE)。
欠拟合与过拟合。
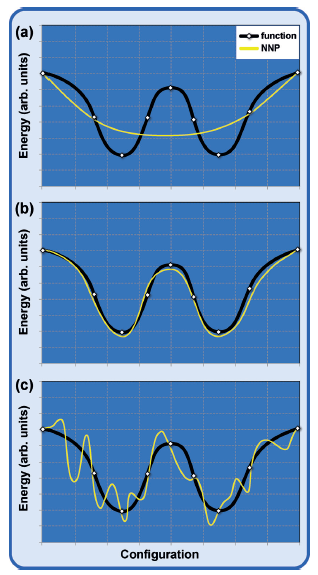
如本节所述,前馈神经网络(FFNNs)已经在许多系统中使用了近20年,这些NNPs受到低维系统的限制。这种限制有几个原因:
- 输入节点的数量与系统的自由度有关,不能任意增加输入节点数量 来阻止神经网络的大小变得难以处理。
- 输入坐标的发展面临极大的挑战,因为系统的能量必须是旋转和平移不变的。此外,必须保证象原子交换的排列对称性。
- 由于训练后的神经网络的输入节点数目不能改变,因此NNP只能用于一个固定的系统大小。
参考文献中有些新的进展......
3.2高维神经网络势能
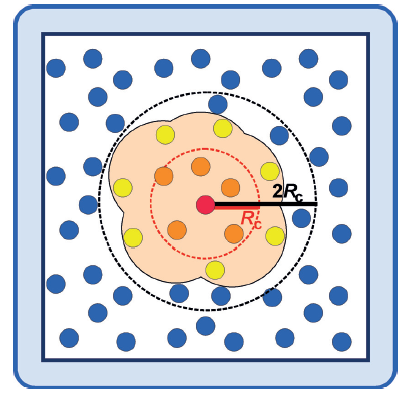
一种适用于包含数千个原子的高维系统的NNP方法;通过对系统中的每个原子使用单独的FFNN(前馈神经网络),克服了上一节讨论的早期NNPs的概念问题。这些“原子NNs”中的每一个都提供了原子m作为化学环境函数的能量贡献。然后,系统中所有Natoms原子的和作为系统的总(短程)能量ES。


对于给定的元素,原子NNs必须具有相同的体系结构(指定隐藏层和神经元的数量)和相同的权重参数。对于包含元素a、B和C的三元系统,生成的HDNNP的结构如图所示。
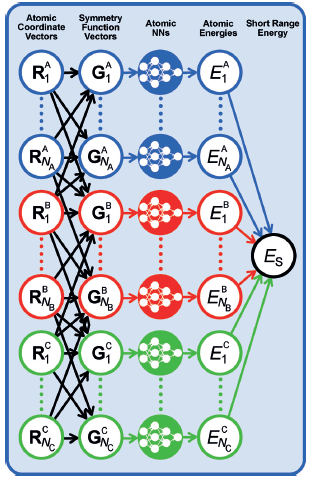
根据系统的化学复杂性,通常使用20到100个对称函数来描述原子环境。
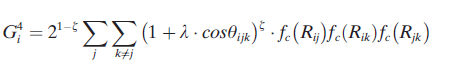

力对原子环境的依赖比原子能量的依赖范围更大(如下图),这是实现能量和力一致的必要条件,对节能分子动力学模拟具有重要意义。
神经网络参数的训练可以使用系统总能量进行,也可以用力来训练。
对于某些系统,例如具有长距离静电相互作用的离子材料,可能需要避免Rc处原子相互作用的截断。这可以通过使用第二组原子NNs显式地添加静电相互作用来实现,与图6中所示的方法相同,来确定与环境有关的原子电荷,从而使总能量描述为:Etot = Es + Eelec 。
HDNNPs具有一些优点:1. 完全满足所有必要的不变量,原子中心对称函数适用于所有类型的系统。甚至定义对称函数形状的特定参数的选择也对特定的系统不敏感。2. 由于原子相互作用的局部性,可以通过使用只包含几百个原子的小系统来训练HDNNPs,并且一旦构建好,就可以应用到更大的系统中。
4.讨论
MLPs的结构非常模块化,本质上由结构描述符和ML方法两大部分组成。HDNNPs利用多体原子中心对称函数[81]来描述原子环境,并利用一组原子NNs将描述向量连接到能量上。对于这两个组件,还有许多其他可能的选择。
目前MLPs面临的另一个挑战是它们对新原子配置的有限可移植性。
MLPs的验证包括以下几个步骤:
1.必须检查感兴趣的结构是否在每个坐标的训练集所张成的描述符范围内。如果检测到外推,则需要扩大PES的有效性范围。
2.在拟合过程中,应尽早停止,避免过拟合。在拟合过程中,对训练集和测试集的误差进行监测,具有最佳泛化属性的拟合是测试集误差最小的拟合,也可以采用更复杂的交叉验证过程。
3.早期停止方法无法识别采样不足的区域(参见图10),因为训练和测试集都来自于可用的参考数据。因此,应该比较几次拟合的预测,只有在很好地一致的情况下,才可以相信其潜力。
5.总结与展望
近年来MLP填补了理论化学和材料科学工具箱中的空白,因为它们可以为复杂材料的反应系统提供非常准确的电位。
ML方法的一个特殊优势是能够以相同的方式描述系统的所有部分,因此不需要预先知道反应的空间位置,而这对于许多系统来说是不可能的。这对于材料科学中的模拟,对于界面上的过程,对于溶液中的反应,特别是质子转移起作用时,尤其有用。
ML方法也有一些突出的缺点,研究进展中付出的巨大努力,需要仔细的验证,大型训练集的需求通常包含成千上万个结构,与训练数据非常不同的配置的有限可转移性,以及目前对包含大约4个元素的系统的限制是有限的(由于配置空间的复杂性呈指数增长,且配置空间中元素的数量也在不断增加)
展望中出现的补充思路(参考文献)。