炎症性肠病和肠易激综合征肠道微生物组组成及功能变化

文献导读
炎症性肠病是一组特定的肠道慢性疾病的统称,主要包括克罗恩病和溃疡性结肠炎—维基百科
肠易激综合征是一组持续或间歇发作,以腹痛,腹胀,排便习惯或大便性状改变为临床表现,而缺乏胃肠道结构和生化异常的肠道功能紊乱性疾病—百度百科
文献信息
- 英文标题:Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome
- 中文标题:炎症性肠病和肠易激综合征肠道微生物组组成及功能变化
- 作者信息:Arnau Vich Vila(第一作者),Rinse. K Weersma(通信作者): Email:[email protected]
- 通信地址:University of Groningen and University Medical Center Groningen, Department of Gastroenterology and Hematology, Groningen, Netherlands.
- 影响因子:16.71 PMID: 30567928 期刊年卷:Sci Transl Med 2018 Dec 19;10(472)
- DOI:10.1126/scitranslmed.aap8914
注:本文章所有图片均在链接里面,想要详细解读图表者建议打开文章最下方链接
文献摘要
肠道微生物组的变化与两种最常见的胃肠疾病,炎症性肠病(IBD)和肠易激综合征(IBS)有关。 这篇文献中,作者使用shotgun metagenomic sequencing对1792名患有IBD和IBS的个体的粪便样品进行病例对照分析。 与对照个体相比,虽然IBD和IBS患者的肠道微生物组之间存在大量重叠,但是通过肠道微生物组组成差异可区分患有IBD的患者和患有IBS的患者。通过将物种水平分布和菌株水平分布与细菌生长速率,代谢功能,抗生素抗性和毒力因子分析相结合,从而确定可能与两种常见胃肠道疾病有关的关键细菌种类。对三个表型良好的荷兰队列的粪便样本进行了shotgun metagenomic sequencing: 总之,作者对1792名参与者的粪便样本进行了分析:355名IBD患者,412名IBS患者和1025名对照者(表S1)。
文献背景
炎症性肠病(IBD)和肠易激综合征(IBS)是两种最常见的胃肠道(GI)疾病。包括克罗恩病(CD)和溃疡性结肠炎(UC)的IBD是以肠炎症为特征的慢性间歇性疾病。 IBS被定义为胃肠道症状的组合,包括腹痛,便秘或腹泻。 IBD和IBS患者有着相似的症状,但IBD的发病机制是由粘膜炎症组成,而IBS的发病机制仍然知之甚少。作者推测肠道微生物组在IBD和IBS中都发挥着重要作用。然而,到目前为止,针对IBD和IBS相关的肠道微生物组测序只能通过低分辨率16S核糖体RNA(rRNA)标记来完成基因测序。并且到目前为止,微生物组的功能性研究仅仅是通过肠道中的单个细菌物种或菌株来进行实现。在这里,作者旨在通过使用高分辨率shotgun metagenomic sequencing 来鉴定完整的肠道微生物组谱并观察来自IBS或IBD个体的粪便样品中的物种水平和菌株水平来弥合之前的16S rRNA测序研究和功能研究之间的差距。作者还通过分析IBS和IBD患者肠道微生物组中的微生物途径,抗生素抗性和毒力因子来确定微生物组靶向治疗的潜在目标。
实验结果
物种水平和菌株水平鉴定显示来自IBD或IBS患者的粪便样品中的微生物组特征
患有CD或UC的患者显示出类似的益生菌肠道微生物组概况。在102个UC相关细菌分类群中,其中87个被发现与CD患者的肠道微生物组谱相关。然而,作者鉴定了15种UC特异性关联,包括Bacteroides uniformis和双歧双歧杆菌。与对照组相比,IBD患者和IBS-GE患者在肠道微生物组中细菌种类的相对丰度增加和减少方面显示出实质性重叠。总共有24个分类群与IBD和IBS相关(表S7和图S2)。这些关联包括几种产丁酸减少的细菌,包括嗜酸杆菌(一种已知的具有抗炎特性的有益细菌,在CD或IBS-GE患者中较低)。与对照组相比,UC患者没有观察到显著差异。除了24个有关联的重叠细菌外,作者还发现了与疾病特异性相关联的细菌。例如,拟杆菌属物种的丰度仅在IBD患者中增加,而在IBS患者中没有增加(表S9)。
在患有IBD或IBS的患者的粪便样品中观察到不同的细菌生长动态
横断面研究提供了在单个时间点细菌分类群的相对丰度的概述,因此不能捕获IBD或IBS患者肠道中微生物生态系统的复杂动态。最近研究已经表明,通过研究肠道细菌基因组中测序读数覆盖率[峰谷比(PTR)]的模式,可以从单个宏基因组样本中推断出细菌生长动态。对于与疾病相关的细菌生长率差异的评估有助于识别活跃生长的细菌,因此可以帮助我们确定与疾病相关细菌分类学结果的优先顺序。在数据集中,可以确定40种物种的细菌生长率,并且与对照组相比(FDR <0.01),在CD患者的菌群中有4种物种有所改变,UC患者有5种,IBS-GE患者有1种,(表S11)。与对照组相比,在患有CD的患者中,脆弱拟杆菌(FDRCD = 0.005)和大肠杆菌(FDRCD = 0.0004)的细菌生长速率增加(表S11)。
肠道微生物组组成可用于区分IBD和IBS-GE
鉴于观察到IBD和IBS-GE患者肠道微生物组的差异,作者研究了使用微生物分类标记作为疾病的潜在预测因子。为了克服缺乏独立复制群组,通过执行10倍交叉验证来估计预测准确性,将疾病群组划分为90%训练集和10%发现集。微生物组合物显示出比目前使用的粪便炎症生物标志物钙卫蛋白更好的预测准确度。当使用微生物分类学数据或微生物途径或两者组合数据集时,仅观察到区分IBD和IBS的能力的微小差异(表S12)。但是当作者将粪便钙卫蛋白测量值与前20个选定的分类法组合时,模型达到最高的预测准确度(具体的请看原文献)。
宏基因组分析揭示了来自IBD和IBS患者的粪便样品中肠道微生物群的功能变化
宏基因组测序能够确定来自CD,UC或IBS-GE患者的肠道微生物组的功能变化。在来自CD,UC或IBS-GE患者的粪便样品中,与对照组相比,许多微生物途径发生了改变。作者确定了微生物功能的重叠和差异,包括氨基酸,神经递质和维生素的合成,以及矿物质吸收的调节和复合碳水化合物的降解(表S15)。在患有IBD和IBS-GE的患者的粪便样品中,丙酮酸盐发酵成丁酸盐(丁酸盐前体)。在患有CD的患者中,发酵途径的减弱,较高的糖降解和醌的合成增加形成了炎症特有的微生物环境(表S15)。在患有UC的患者中,产生丁酸盐和乙酸盐的途径减少,并且产生乳酸的途径增加。然而,在IBS-GE患者中,代谢的特征在于发酵增加和碳水化合物降解途径 。作者发现几种微生物l-精氨酸途径的改变,表明CD患者可能存在L-精氨酸的消耗。
IBD或IBS患者的肠道微生物组中毒力因子的丰度增加
毒力因子通过以下几种机制促成细菌的致病潜力,即增加细菌与肠粘膜的粘附,免疫系统逃避或抑制宿主免疫应答。作者评估了宏基因组读数与来自毒力因子数据库(VFDB)的蛋白质序列之间的同源性。在CD,UC,IBS-GE或IBS-POP患者中,与对照组相比,262种毒力因子的相对丰度增加(FDR <0.01;表S16)。在患有CD的患者中,216种毒力因子的丰度增加(表S16)。肠杆菌素的丰度与肠杆菌的相对丰度相关。毒力因子的丰度增加也反映在CD患者中肠杆菌素途径的增加(ENTBACSYN_PWY,FDR = 0.006;表S15)。许多病原体已获得有效的铁摄取机制,使其在低铁环境中具有生存优势。这反映在CD患者中几种微生物铁摄取途径的改变。在UC患者中,35种毒力因子增加,例如,MU-毒素及其含有nagI,nagJ和nagL的转运蛋白复合物的相对丰度增加(FDRnagI = 3.56×10-5,FDRnagJ = 4.59×10- 13,FDRnagL = 9.11×10-9;表S16)。
IBD和IBS患者的微生物组组成的变化对抗生素抗性负荷有影响
宏基因组测序为大规模研究IBD或IBS患者的抗体组提供了机会。为了观察IBD或IBS患者的肠道微生物组中是否存在抗生素耐药性的增加,作者评估了来自抗生素抗性数据库,综合抗生素抗性数据库(CARD)的宏基因组读数和蛋白质序列之间的同源性。随后,为了确定可能含有抗生素抗性蛋白的微生物,作者将抗生素抗性基因的丰度与分类丰度相关联。在CD患者中,编码抗生素抗性蛋白的142个基因的丰度高于对照组。在这些抗生素抗性蛋白中,63个基因是外排复合物的组分,其从细菌中除去抗生素,从而防止抗生素有效地起作用(表S17)。这些外排复合物由跨越内膜,周质和细菌外膜的三种蛋白质组成。一些外排泵只能输送一种特定类型的抗生素,而其他外排泵,称为多药外排泵,可以输送几种类型的抗生素。 CD患者中增加的抗生素抗性蛋白TolC是包含几种多药外排泵的外膜蛋白。CD患者中TolC的丰度与埃希氏菌属的分类丰度相关。在UC患者中,编码抗生素抗性蛋白的66个基因的丰度高于对照组。抗生素抗性基因cepA的丰度与拟杆菌属的丰度相关,其在UC和CD患者中增加(Spearman系数,rho = 0.86; FDR <1.0×10-16;表S17)。编码抗生素抗性蛋白的几个基因在IBS患者中增加,并且与对照相比,IBS-GE患者中32个抗生素抗性基因的丰度增加。IBS-GE患者中增加最多的抗生素耐药蛋白之一是mecB(FDR = 0.0001;表S17),其参与对甲氧西林的抗性。 这种蛋白质通常存在于属于大果属(Macrococcus)的物种中,与金黄色葡萄球菌属(Staphylococcus genus)密切相关。 在IBS-POP患者中,与对照相比,编码抗生素抗性蛋白的13种基因的丰度增加。 通常在肺炎链球菌中发现的PBP2x与我们的肠道微生物组数据中链球菌属的分类丰度(Spearman系数,rho = 0.91; FDR <1.0×10-16;表S17)高度相关。
肠道微生物组的变化与疾病特异性因子和疾病亚型有关
先前的研究已经确定肠道微生物组的组成受到超过100种内在和外在因素(例如,饮食因素,药物,疾病和人体测量因子因素)的影响。然而,在IBD和IBS中,可以改变肠道微生物组组成和各种表型(例如,排便频率,药物使用和先前进行的GI外科手术干预)。因此,作者重新计算了内在和外在因素与总体微生物组成(Bray-Curtis相似性),α多样性(Shannon指数)和基因丰富度之间的关系。这些结果连同内在和外在因素的相关性(表S22至S25)导致了随后的关联分析中包括的因子列表(表S26)。在分类学(表S27至S34)和微生物途径(表S35至S43)上进行单变量和多变量的病例内关联分析。在CD中,只有1%的微生物变异可以通过炎性疾病活动来解释(FDR = 0.077;表S18)。相比之下,CD患者的回盲切除术导致回盲瓣的切除是解释5%方差的因素(FDR = 0.00159;表S18)。回盲瓣的缺失与微生物和基因丰富度的降低有关,特别是有益菌F. prausnitzii(FDRCD-ileal = 8.01×10-10;表S27)和Ruminococcaceae家族(FDRCD-ileal =)的减少。 4.63×10-10;表S27)和梭杆菌的增加(FDRCD-ileal = 0.002;表S27)。这表明去除回盲瓣对IBD患者的肠道微生物组具有负面影响。 CD患者补充维生素D与Akkermansia muciniphila(FDRCD = 0.19;表S27)的丰度减少有关,这是一种在低纤维环境中生长的粘蛋白降解细菌。
讨论
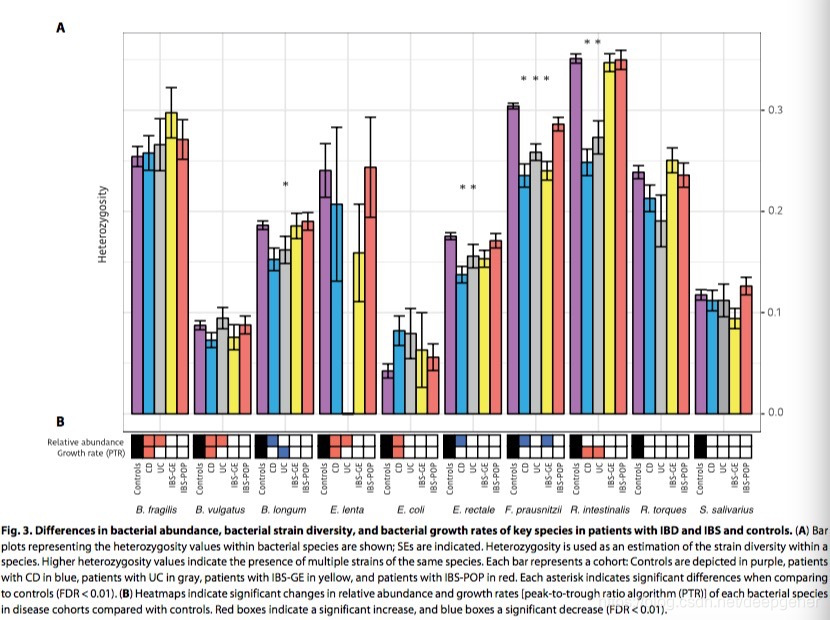
本文亮点:使用shotgun metagenomic sequencing 数据使我们能够以高分辨率探索肠道微生物生态系统的复杂性。我们还能够描述微生物群落的一些重要特征,包括菌株多样性,生长动态,以及参与细菌毒力和抗生素抗性机制的基因的存在,这些机制可以为机会性和病原性微生物提供适应性优势。作者还探讨了微生物途径谱的变化,提供了有关微生物组生态失调的功能后果的相关信息。这些数据集的整合使我们能够确定关键物种作为IBD和IBS功能研究的目标(图3),并将IBD和IBS的病因学和发病机制的知识与肠道微生物组联系起来,为治疗提供潜在的新靶点。
不足点:在将研究的结果转化为临床实践之前,需要更多的额外证据来克服我们研究的局限性。在本研究中,作者使用了两组患者人群,这些患者已经诊断出患有IBD或IBS。因此,预测模型并未反映临床情况,即未接受治疗的患者或患有其他合并症的患者可能存在不同的微生物组特征。此外,实验室方案,测序技术或样品的地理来源的变化也可能影响我们模型的准确性。患有既定疾病的患者的横断面队列使我们能够发现许多不同亚型的影响;然而,这些队列只能提供有关IBD或IBS发病机制的有限信息。纵向研究将有助于确定疾病的动态,以及区分因果症和疾病后果的微生物特征。该研究的另一个局限是明确定义的IBS患者数量相对较少。因此,作者无法对IBS亚型进行深入表征,如便秘或腹泻患者。在此研究中,每个参与者都可以获得许多表型特征,例如药物使用或生活方式,这使作者能够在考虑重要的混杂因素的同时进行严格的病例对照分析。在疾病背景下研究微生物组时,使用特征明确的队列应该成为一种常见的做法。使用诸如质子泵抑制剂或缓泻剂的药物,其经常被IBD或IBS患者使用,对肠道微生物组组成具有很大影响。
附加点:疾病活动解释了UC患者微生物组成的大部分变异,而疾病定位和肠切除对CD患者的肠道生态系统有很大影响。这一事实强调了在分析IBD或IBS患者的微生物组成时收集和考虑疾病特异性表型的重要性。在患有IBD的患者中观察到肠道微生物组的生理障碍。 IBD的两个主要亚型(CD和UC)在其肠道微生物特征中显示出显著的重叠。通过探索IBD和IBS中肠道微生物群组的致病潜力,我们能够识别UC患者中的其他潜在目标,如Mu毒素。尽管这些发现仍需要通过靶向方法或转录组学分析进行验证,但我们提出的毒力因子关联可以更好地理解这两种疾病的发病机制。我们在IBD和IBS患者的肠道微生物组组成和功能潜力中发现的变化可能会产生新的方法,从而有助于临床实践中的诊断。 本文研究结果表明,将来使用针对关键细菌物种的探针可以补充粪便钙卫蛋白测量项,以区分IBS和IBD的诊断。
参考文献
[1]点击查看原文献
[2]点击查看文献补充图
深度基因小伙伴温馨提示:
- 如果我们对文章理解有偏差,非常欢迎大家向我们反馈,我们会认真阅读建议并修改,另外有意愿加入我们的小团队的老师和同学可发送邮件至我们的邮箱:[email protected] 祝大家科研顺利,生活开心!
- 想要了解更多内容请访问我们的深度基因网站:http://deepgener.wordpress.com/
- 点击查看上一篇文章