如何计算核酸多样性 Pi
本期笔记分享关于核酸多样性pi计算的方法和相关技巧,主要包括原始数据整理、分组文件设置、计算原理、操作流程、可视化绘图等步骤。
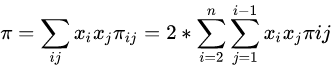
基因组Pi核酸多样性(Pi nucleic acid diversity)是一种遗传学研究中用来描述种群内核酸序列多样性的指标,通常用π(pi)符号表示。
它衡量了在一组生物个体的基因组中,不同核苷酸位置上的多态性水平,这种多样性是通过比较不同个体之间的DNA或RNA序列来测定的。
需要输入什么样的数据?
-
样品分组数据
一般在计算pi的时候,需要根据不同亚群或分组来计算,比如高个子和矮个子分成两组,或者按照表性差异、起源地点等分成若干组。
需要将组的样品ID保存为一个txt文档,每行放一个样品ID,不同组的样品放在不同txt文件中。
cat pop1.txt
sample001
sample002
sample006
...
cat pop2.txt
sample012
sample054
sample269
-
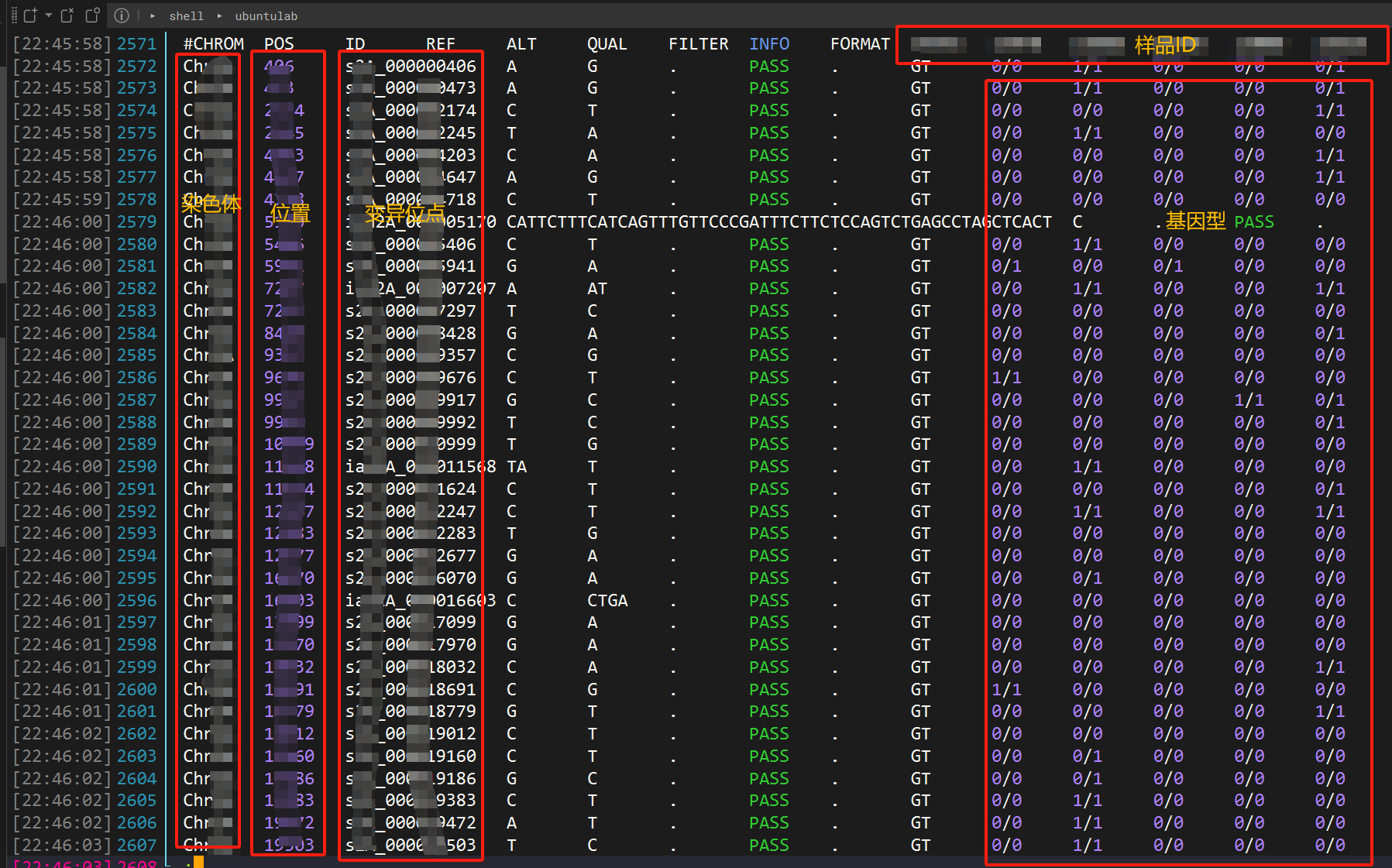
基因型数据
需要的基因型数据是VCF格式(常规存储变异信息的一种格式,通常由测序上游分析GATK标准流程获得),其中包含了区间内每个SNP在每个样品的基因型。
如何进行计算?
# 首先加载所需要的R包
library(tidyverse)
library(vcfR)
library(scales)
# 备注:请提前安装vcftools并添加到环境变量
设置参数
# VCF <- "xxx.vcf"
# QTL <- "Gene1"
# SNP <- "MY_SNP_ID"
# mycol <- c("#eb6f51","#20b694","#3560a3","#f8c06f","#004b1c","#5c2d91")
mycol参数可以设置想要的颜色,最终绘图时会根据此参数调整折现颜色。VCF、QTL、SNP根据实际情况进行修改。
group_list <- c("pop1","pop2","pop...")
group_list列表存储亚群或者分组信息,每个亚群分别计算pi然后再合并绘图。
这里可以用任意分组规则,只要能把样品分成一堆一堆就可以,目的是为了研究不同分组之间的差异,还有同一组内的变化趋势。
计算Pi核酸多样性
for (i in group_list){
shell_cmd <- str_c("vcftools",
"--vcf",VCF,
"--keep",str_c(i,".txt"), # 这里会自动读取每个分组文件
"--window-pi","100000", # 100kb窗口
"--window-pi-step","10000", # 10kb步长
"--out",str_c("out_pi/",i),
"",sep = " ")
print(shell_cmd)
system(shell_cmd)
}
这段R语言代码是一个循环,用于执行一系列的系统命令。以下是逐步解释:
for (i in group_list):
遍历名为group_list的列表中的每个元素,将当前元素赋值给变量i,然后执行循环体中的操作。
shell_cmd <- str_c(...):
在每次循环中,创建一个字符串变量shell_cmd,其中包括一系列参数依次进行调用,自动生成命令代码。
print(shell_cmd):
打印当前循环中生成的shell_cmd字符串,以便查看每次循环生成的命令。
system(shell_cmd):
使用system函数执行生成的shell_cmd命令,这将在系统上运行相应的vcftools命令,执行一些与VCF文件处理相关的任务,如计算窗口内的π值。
总之,这段代码的作用是循环遍历group_list中的元素,每次循环生成一个用于运行vcftools命令的字符串shell_cmd,然后执行该命令。
结果可视化
以下R语言代码的目的是创建一个包含数据框(data frame)的列表,并将一些数据加载到这些数据框中,最后将它们合并成一个大的数据框,用于ggplot绘图。
df_plot <- list()
# 创建空列表,并读取数据
for (i in group_list){
df_plot[[i]] <- read_table(str_c("out_pi/",i,".windowed.pi"))
df_plot[[i]][,2] <- df_plot[[i]][,2]/1000000 # 转换物理位置为MB
df_plot[[i]][,6] <- i
colnames(df_plot[[i]])[6] <- "Group"
}
df_plot_all <- bind_rows(df_plot)
以下是每行代码的具体解释信息:
df_plot <- list():首先创建一个空列表df_plot,用于存储数据框。
for (i in group_list):通过for循环遍历group_list中的元素,其中i代表当前循环的元素。
df_plot[[i]] <- read_table(...):在每次循环中,创建一个数据框,并将其存储在df_plot列表中的以i命名的位置。read_table函数用于读取数据文件,文件路径由str_c("out_pi/",i,".windowed.pi")构建,i是当前group_list中的元素。这个数据框包含了从指定文件中读取的数据。
df_plot[[i]][,2] <- df_plot[[i]][,2]/1000000:将df_plot列表中的第i个数据框的第2列数据(可能是物理位置)除以1000000,将其转换为兆字节(MB)。
df_plot[[i]][,6] <- i:在第i个数据框中添加一列,该列的所有值都设置为当前循环的i值,这列通常用于标识数据来源的组。
colnames(df_plot[[i]])[6] <- "Group":将第i个数据框的第6列的列名更改为"Group",以便更清晰地表示这一列的含义。
df_plot_all <- bind_rows(df_plot):最后,使用bind_rows函数将df_plot列表中的所有数据框合并成一个大的数据框df_plot_all,这个数据框包含了来自不同组的数据,每个组的数据位于不同的行中,且具有"Group"列来标识它们所属的组。
绘图
下面这段代码使用ggplot2包创建一个散点拟合曲线图并将图形保存为PDF文件。
p1 <- ggplot(df_plot_all)+
geom_smooth(aes(BIN_START,PI,color=Group),
method = "loess",span = 0.1 ,se = F,linewidth = 1 )+
# geom_line(aes(BIN_START,PI,color=Group),linewidth=1)+
scale_color_manual(values = mycol)+
xlab(str_c("Physical Postion "))+
ylab("Pi")+
ylim(0,0.01)+
theme_bw()+
theme(legend.position = "none")
ggsave(filename = str_c("out_plot/pi_10MB/",QTL,"_",SNP,"_pi_BG_100kwindow_10kstep.pdf"),
plot = p1,width = 8,height = 4)
以下是每行代码的详细解释:
p1 <- ggplot(df_plot_all) +:创建一个ggplot2的图形对象,基于数据框df_plot_all。
geom_smooth(aes(BIN_START, PI, color = Group), method = "loess", span = 0.1, se = F, linewidth = 1):在图形上添加平滑曲线。使用BIN_START列作为x轴,PI列作为y轴,根据不同的Group组别进行着色。曲线平滑方法为loess,span参数控制平滑度,se = F表示不要显示置信区间,linewidth = 1设置曲线的线宽。
scale_color_manual(values = mycol):自定义颜色映射,将mycol中的颜色赋予不同的组别。
xlab(str_c("Physical Position"):设置x轴标签为"Physical Position"。
ylab("Pi"):设置y轴标签为"Pi"。
ylim(0, 0.01):限制y轴的范围,从0到0.01。
theme_bw():应用白底主题,使图形背景为白色。
theme(legend.position = "none"):隐藏图例,因为legend.position设置为"none"。
ggsave(filename = str_c("out_plot/pi_10MB/",QTL,"_",SNP,"_pi_BG_100kwindow_10kstep.pdf"), plot = p1, width = 8, height = 4):最后一行代码保存图形为PDF文件。文件名根据变量QTL和SNP动态生成,保存在指定路径下
以上就是计算核酸多样性并可视化的方法,欢迎您看到这里,如果感觉有用请转发收藏,以备不时之需。
本文由 mdnice 多平台发布