2023年4月,南方科技大学夏雨研究团队在《Genome Research》上发表题为“Genome enrichment of rare and unknown species from complicated microbiomes by nanopore selective sequencing”的研究论文。利用Oxford Nanopore适应性采样技术,通过一段短时间的正常测序,确定出群落的大致结构和高丰度物种的组成,以形成进行选择性测序所需的参考序列。将metaRUpore应用于高温厌氧反应器(thermophilic anaerobic digester, TAD)群落和人类肠道微生物群落,metaRUpore成功将测序通量从高丰度种群转移到稀有物种,并促进了稀有物种的高质量基因组(high-quality metagenome-assembled genomes, HQ-MAGs)的组装。该方法简捷且稳定,适用于具有中等计算资源的实验室,并有可能成为未来对复杂微生物群落进行宏基因组纳米孔测序的标准方法。

DOI:10.1101/gr.277266.122
文章摘要
稀有物种是微生物群落中的重要成员,但由于其丰度低,很难被组装成完整的基因组。ReadUntil(RU)策略使纳米孔测序仪能够实时地对特定DNA分子进行选择性测序,这为富集稀有物种提供了潜在的方法。尽管通过RU弹出具有已知基因组序列的宿主DNA(例如人类宿主DNA)来富集稀有物种的方法已被广泛应用,但在群落组成不明的环境样品中,基于RU的稀有物种富集仍然存在难题,这是由于在这些样品中许多物种缺乏相应的参考基因组序列。因此,本文提出了metaRUpore来解决这个问题。MetaRUpore通过一段短时间的正常测序,确定出群落的大致结构和高丰度物种的组成,以形成进行选择性测序所需的参考序列。本研究将metaRUpore应用于高温厌氧反应器(thermophilic anaerobic digester, TAD)群落和人类肠道微生物群落,metaRUpore成功将测序通量从高丰度种群转移到稀有物种,并促进了稀有物种的高质量基因组(high-quality metagenome-assembled genomes, HQ-MAGs)的组装。该方法简捷且稳定,适用于具有中等计算资源的实验室,并有可能成为未来对复杂微生物群落进行宏基因组纳米孔测序的标准方法。
研究背景
微生物群落由大量的稀有微生物组成,它们对于保持微生物生态系统的健康和稳定发挥着至关重要的作用。例如,氨氧化细菌或古菌(AOB/AOA)和ANAMMOX,这些低丰度且生长速率缓慢的自养微生物承担着自然界中氮循环的限速步骤。因此,认识这些稀有微生物的功能对于理解微生物群落的动态变化和生态功能至关重要。
高通量宏基因组全基因组测序开启了一个新的时代,从此我们能够从宏基因组测序数据中组装基因组草图,以了解微生物群落中占大多数的不可培养的微生物的生态和进化特征。然而,想要组装出高质量(通常定义为>90%的完整度,<5%的污染度以及含有完整的rRNA操纵子)的低丰度微生物的MAGs总是很困难。在宏基因组测序中,低丰度微生物的测序覆盖率往往很低,甚至会被直接忽略。若要使低丰度物种达到足够的基因组覆盖率,通常需要进行极深的测序。如果研究的目的只集中在稀有微生物,那将是对测序通量的极大浪费;数据分析过程也会变得同样棘手,因为将未知的基因组从数百 GB到 TB 的数据中组装出来是一项巨大的计算挑战。
为了从高丰度背景中提高稀有类群的覆盖率,一些基于分子生物学的方法被提出,如在文库制备中采用杂交捕获或通过CRISPR-Cas9来富集目标、通过皂苷消耗人的DNA用于快速临床诊断低丰度的病原菌等。然而,这些方法需要使用额外的试剂和准备程序。更重要的是,他们需要已知的富集或消耗目标才能设计实验,这样的方法似乎并不适用于富集组分未知的宏基因组群落中的低丰度物种。
与上述在测序之前进行的基于实验的方法不同,纳米孔测序(Oxford Nanopore Technology)的用户可以对测序系统进行编程,在测序的同时反转纳米孔两侧的电压以弹出不感兴趣的DNA片段,这为富集宏基因组中的稀有微生物提供了潜在的解决方案。这种“选择性测序”或ReadUntil (RU)的策略最早由Matthew Loose及其同事于2016年提出。最先采用的RU方法使基于动态时间扭曲(DTW)算法来比对测序原始信号和DNA参考序列,但是该算法需要大量的计算资源,DNA参考序列只能限制在数百万个碱基之内,这限制了其广泛使用。UNCALLED工具同样采用了将原始信号与DNA参考序列比对的方法,其计算量比DTW小,但仍然需要大量的计算资源。最近提出的Readfish工具摒弃了复杂的原始信号比对的算法,它利用现有的Oxford Nanopore工具来设计和控制选择性测序实验。到目前为止,RU的应用主要限于消除已知宿主物种或富集已知目标,例如吸血昆虫的有丝分裂基因组等。
通过选择性地测序低丰度物种的DNA,同时弹出高丰度物种的DNA,选择性测序为富集宏基因组样本中的稀有微生物提供了潜在的解决方案。尽管如此,通过选择性测序富集真实的环境样本中的低丰度微生物仍然具有挑战性,因为其群落组成是未知的,并且其中的大部分物种在公共数据库中没用相应的参考基因组。为了实现在复杂环境微生物组中有效富集稀有物种,我们设计了metaRUpore,它由选择性纳米孔测序和一些必要的生物信息学脚本组成,可在复杂环境微生物组中富集低丰度物种。本文通过实施metaRUpore这一新方法,来富集多样的样本中的低丰度微生物,并推进我们对复杂生态系统中微生物多样性和生态动力学的理解。

研究结果
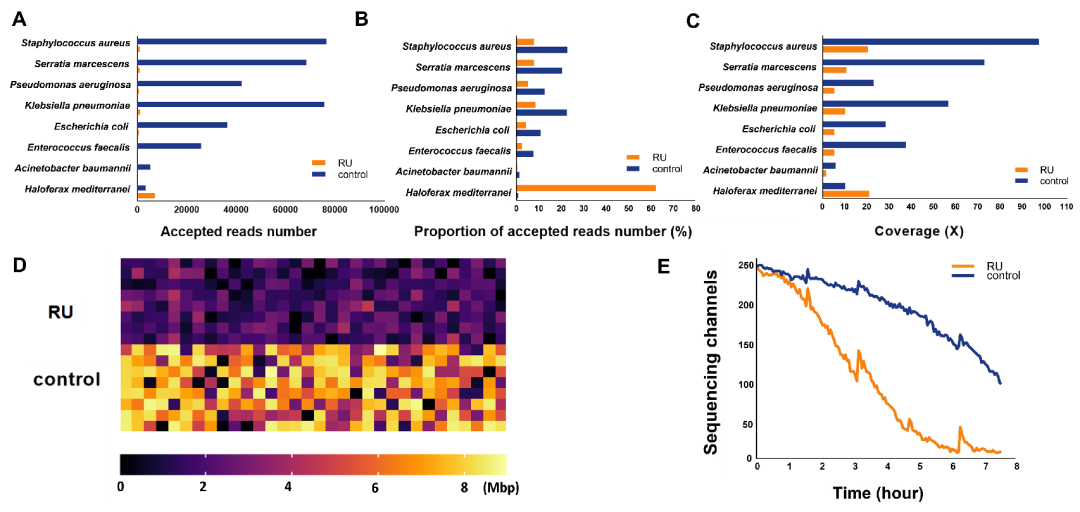
Fig1.通过RU富集模拟群落中的低丰度物种。(A)RU和对照组中八种微生物的被接受的测序片段数条形图。(B)RU和对照组中八种微生物的被接受的测序片段比例的条形图。(C)RU和对照组中八种微生物基因组覆盖率的条形图。(D)RU和对照组测序进行中每个测序通道的数据产量热图(E)RU和对照组测序进行测序通道数量图
首先构建了一个由八种微生物组成的模拟群落。在该模拟群落包括占1%的噬盐古菌(Haloferax mediterranei)和其他七种细菌。使用Readfish选择性测序工具包在GridION X5测序平台对该模拟群落进行富集古菌的纳米孔选择性测序。设置MinION芯片的一半通道进行纳米孔选择性测序以富集相对稀有的古菌,另一半进行正常测序作为对照。在选择性测序通道中,大于99.9%的嗜盐古菌的DNA片段被保留,而大于99%的细菌片段被弹出。相较于正常测序方法,嗜盐古菌在群落中由原本较为稀有的物种变成了占据优势地位的物种,其覆盖率也增加了两倍(图1bc)。尽管选择性测序在模拟群落中的应用达到了相当理想的对稀有物种的富集效果,但在富集目标比例为1%时,进行选择性测序的一半通道的数据产量比普通测序通道的产量低约60%(图1d),这提示在纳米孔选择性测序中要设置合理的弹出目标,以实现富集效率和通量损失之间的最佳平衡。
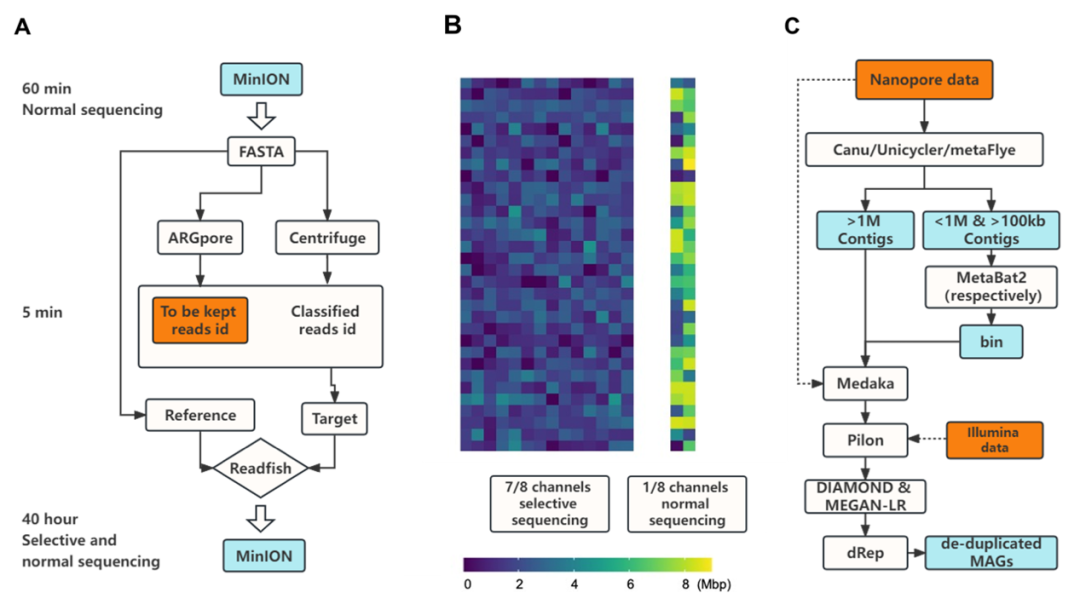
图2.(A) metaRUpore 的工作流程。(B)在metaRUpore中,MinION芯片被配置为两部分,1/8测序通道用于正常测序,其余通道用于选择性测序。(C)组装HQ-MAG的生物信息学流程。
metaRUpore的工作流程可以分为三个连续的步骤:首先进行短时间的正常测序,以获取整个群落的总体情况和优势种群的信息;然后进行生物信息学分析,以确定选择性测序所需的参考和目标数据集;最后进行40小时的选择性测序,以富集样品中的稀有种群(图2ab)。其中生物信息学分析流程可以在5分钟内完成,以快速生成确定的参考序列和目标数据集,对随后进行后续的选择性测序工作将不产生显著影响。同时,我们建立了有效的组装策略(图2d)。
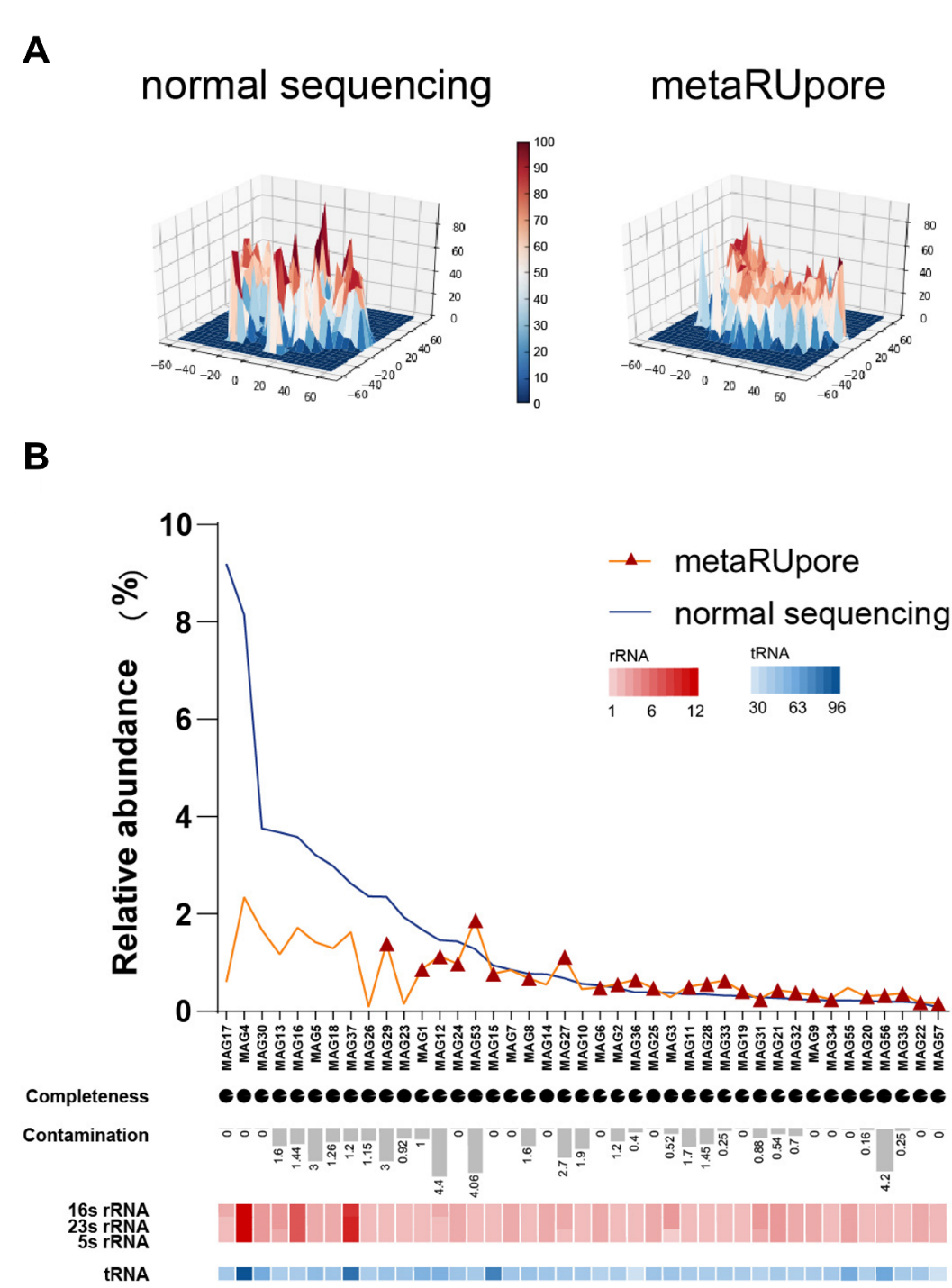
图3. (A) TAD群落正常测序数据集和通过metaRUpore进行选择性测序产生的数据集的四碱基频率的t-SNE降维结果的3D密度图(B) 41个组装出的HQ-MAGs在正常和RU测序数据集中的分布。红色三角形表示仅能由metaRUpore数据组装得到而不能由正常测序数据集组装出的HQ-MAGs。饼图和条形图代表基因组的SCG完整性和污染度水平。16S rRNA、23S rRNA和5S rRNA基因的拷贝数由红色热图表示,而 tRNA 的拷贝数由蓝色热图表示。
对比用正常测序数据和通过metaRUpore进行选择性测序产生的数据做出的3D密度图(图3a),可观察到群落结构的显著变化:正常测序3D密度图有许多密度高峰;而选择性测序3D密度图中这些高峰的数量明显减少。这表明TAD群落中高丰度物种的DNA在通过metaRUpore进行选择性测序后被有效地弹出,不同物种的覆盖变得更接近,群落变得更加均一。从通过metaRUpore进行选择性测序获得的数据中组装了46个DQ-MAGs,其中有41个HQ-MAGs,这些HQ-MAGs中有6个MAGs是完整的环状宏基因组组装基因组。所有的HQ-MAGs包含的contigs数都不超过13个,平均N50>2Mbp,是高度连续的。相比之下,正常测序数据集产生了29个DQ-MAGs,其中包括16个HQ-MAGs,这16个MAGs中绝大多数MAGs也能由用metaRUpore进行选择性测序的数据组装得到,而只能由metaRUpore数据组装得到而不能由正常测序数据集组装出的26个HQ-MAGs,主要由TAD群落的稀有物种组成,这表明metaRUpore促进了更多稀有物种基因组的组装。
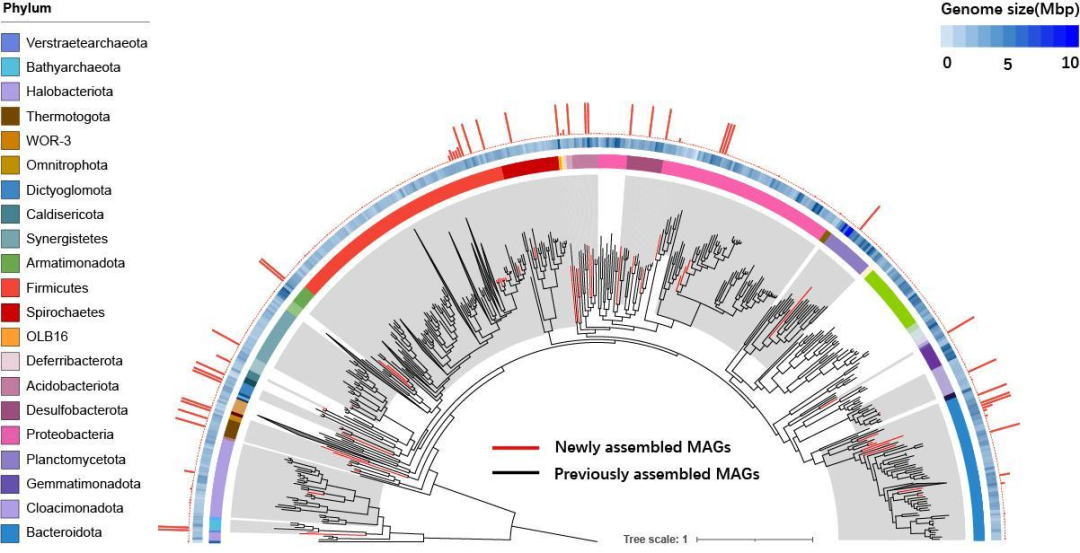
图4: 由metaRUpore数据组装的的41个HQ-MAGs(红色分支)和全球基因组参考数据库中的1108个HQ-MAGs(黑色分支)构建的系统发育树
树外圈由内到外分别代表:(1)MAGs的门(2)基因组大小(3)基因组连续性(连续性定义为contigs数量的倒数);灰色阴影区域表示由metaRUpore数据获得的基因组接近完整的门
这41个HQ-MAGs给厌氧反应器群落全球基因组参考数据添加了5个新门(OLB16、WOR-3、Omnitrophota、Gemmatimonadota和Deferribacterota)和21个门的第一个HQ-MAG。通过metaRUpore,属于产甲烷古菌门Bathyarchaeota和Verstraetearchaeota的低丰度物种的测序通量被大大提高,并被组装成接近完整的高质量的基因组。
通讯作者介绍

夏雨博士
南方科技大学,环境科学与工程学院,副研究员,博士生导师
环境微生物与生态基因组学实验室负责人(XYLab)。研究兴趣集中于:结合BONCAT等先进分子生物学手段以及Nanopore测序为代表的高通量组学技术,解密生物处理反应器、极端自然系统及洁净室内环境中微生物群系的种间互作关系以及关键基因(耐药基因)转移规律。近五年来在Environmental Science & Technology, Water Research,Genome Research等领域顶级期刊发表论文50余篇,总引用次数 3000余次(Google Scholar), 作为第一发明人申请发明专利2项。现任Frontiers in Environmental Science副主编,iMeta期刊执行副主编,中国工程院院刊Engineering青年编委。担任国自然面上项目、青年项目负责人及科技部重点研发计划课题负责人。曾担任南方科技大学教授委员会代表委员,美国微生物协会香港地区青年大使。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读