参考论文

摘要
面部形态——人类外表的一个显著特征是高度可遗传的。先前关于面部形态遗传基础的研究主要在欧洲血统队列(EUR)中进行。将数据驱动的表型和多变量全基因组扫描方案应用于东亚血统个体(EAS)的大量三维面部图像,我们在166个基因座(62个新基因座)中确定了244个与典型范围面部变异相关的变异。一项新提出的多基因形状分析表明,变异对EAS面部形状的影响可以推广到EUR。在此基础上,我们进一步确定了13种与EUR和EAS人群面部形状差异有关的变异。进化分析表明,EUR和EAS种群鼻子形状的差异是由定向选择引起的,主要是由于欧洲人的局部适应。我们的研究结果说明了不同人群面部差异的潜在遗传基础。
背景概念
基因座:
基因座是指基因组中的特定位置,也被称为基因位点或基因位置。基因座可以是基因或其他DNA序列的位置,这些位置在不同个体之间可能会发生变异。在一个基因座上,一个个体可以有不同的基因型。基因型是指一个个体在某个基因座上的基因组合。例如,对于人类的血型基因座,可能有A型、B型、AB型和O型等不同的基因型。
单核苷酸多态性(SNP):
单核苷酸多态性(Single Nucleotide Polymorphism,SNP)是指基因组中单个核苷酸的变异,是人类遗传变异的最常见形式之一。SNP通常出现在DNA序列的不同位置,其中一个核苷酸替换为另一个核苷酸,如A、C、G或T。SNP的存在可以影响一个基因的表达或功能,可能与一些遗传病和复杂疾病的发生有关。SNP通常是一种常见的变异形式,其在人类种群中的频率通常大于1%。SNP的多样性是由于在人类漫长的进化过程中,人类遭受了许多不同的生态和环境压力,从而导致了不同基因型的选择。在研究中,SNP通常用于进行基因关联研究,以确定其与特定疾病或表型的关联性。SNP的基因关联研究是通过对某个基因或某个基因区域中的SNP进行分析,找到与某种疾病或表型相关的SNP。这些SNP通常与某些生物学过程相关,如基因表达、蛋白质功能等。因为SNP是遗传变异的常见形式,它们已成为基因组研究中最常用的标记之一。在基因组学和生物医学研究中,SNP被广泛应用于基因组关联研究、基因型鉴定、群体遗传学、进化研究等领域。
基因关联研究(GWAS):
基因关联研究(Genome-Wide Association Study,GWAS)是一种通过对大量单核苷酸多态性(SNP)进行全基因组扫描来研究基因与疾病或其他复杂表型之间关系的方法。GWAS是一种无偏的方法,可以在不需要假设或预先知道特定基因或途径的情况下,快速发现与人类疾病相关的基因和基因区域。在GWAS中,研究人员通常会对两个或多个群体进行比较,其中一个群体患有某种疾病或表型,而另一个群体则不患有该疾病或表型。研究人员使用高通量基因芯片或测序技术来分析这些个体的基因组,并检测每个个体的SNP。然后,研究人员将每个SNP与疾病或表型之间的关联性进行比较。如果一个SNP在患有该疾病或表型的人群中更为普遍,则认为该SNP与该疾病或表型之间存在显著关联。GWAS已被广泛应用于许多疾病的研究,如心血管疾病、糖尿病、癌症、自闭症等。它已经产生了许多重要的研究发现,包括发现新的遗传变异与疾病的关联、确定新的疾病途径和发现新的靶标。同时,GWAS也存在一些局限性,例如,它只能检测到单个SNP与疾病之间的关联,而不能检测到多基因相互作用和环境因素等其他复杂的影响。
genome-wide significant threshold (全基因组显著性阈值):
全基因组显着性阈值设置在非常严格的水平,例如 p < 5 × 10^-8(或 0.00000005)。这意味着只有 p 值低于此阈值的关联才被认为具有统计学意义,并且可能代表遗传变异与正在研究的性状或疾病之间的真实关联。
设置全基因组显着性阈值有助于确保只有此阈值的选择基于 Bonferroni 校正,它调整 p 值阈值以考虑多重假设检验。Bonferroni 校正将所需的显着性水平(通常为 0.05)除以执行的独立测试的数量,从而产生更严格的阈值来控制增加的误报机会。最强和最强大的关联被认为是显着的,从而减少由于 GWAS 中的偶然发现而报告假阳性的可能性。重要的是要注意,此阈值不能保证已识别关联的生物学或临床相关性,而是作为在 GWAS 背景下宣布显着结果的统计标准。
minor allele frequency(次要等位基因频率) :
用于描述人群中特定遗传变异中不太常见的等位基因的频率。MAF生物学意义
-
遗传多样性:更高的 MAF 表明种群内更大的遗传多样性。高 MAF 表明该变异相对常见并且存在于人群中较大比例的个体中,而低 MAF 表明该变异不太常见并且存在于较小比例的个体中。
-
自然选择:MAF 可以提供有关作用于遗传变异的进化力的信息。如果一个变体的 MAF 非常低,则可能表明它处于负选择状态,这意味着它由于其有害影响而被选择性地从种群中移除。相反,高 MAF 可能表明正选择,表明该变体具有某些有利性状或选择优势。
-
性状或疾病相关性:在遗传研究的背景下,MAF 还可以影响变异与特定性状或疾病的相关性。具有较高 MAF 的常见变异更有可能得到广泛研究,并且可能与各种性状或疾病有关。具有较低 MAF 的罕见变异可能对研究罕见遗传疾病或研究具有独特遗传特征的特定人群感兴趣。
-
统计能力:MAF 可以影响检测遗传变异与性状或疾病之间关联的统计能力。具有更高 MAF 的变体更有可能具有更大的样本量,从而为关联研究提供更多的统计能力。相反,具有较低 MAF 的罕见变异通常需要更大的样本量或替代研究设计才能获得足够的检测能力。
多变量全基因组扫描协议:
多变量全基因组扫描协议是一种针对多个表型特征同时进行基因关联研究的方法。与传统的单一表型GWAS相比,多变量全基因组扫描协议可以同时考虑多个表型特征,从而更全面地评估基因与复杂表型之间的关系。
多变量全基因组扫描协议的实施包括以下几个步骤:1)确定研究的表型特征:在多变量全基因组扫描中,需要选择多个表型特征进行研究,例如身高、体重、血压、血糖等。这些表型特征应该与研究问题相关,且需要有充分的测量数据。2)数据预处理:在多变量全基因组扫描中,需要对原始数据进行预处理,以去除可能对结果产生干扰的噪声或偏差。预处理步骤通常包括数据清洗、归一化、缺失值填充等。3)基因型数据准备:需要将每个个体的基因型数据与其表型特征数据相对应,以建立数据集。通常采用SNP芯片或基因组测序技术来测量每个个体的基因型数据。4)基因关联分析:进行基因关联分析来确定每个SNP与表型特征之间的关系。
本文的数据集:分为3个队列
NSPT队列(n=3322):广西南宁(n=1326);江苏省泰州市(n=986);河南省郑州市(n=1010)。
NHC队列(n=4767):河北省唐山市
(以上两个队列构成了发现数据集)
TZL队列(n=2881):江苏泰州(构成了复制数据集)
研究结论
GWAS on facial phenotypes discovered 244 leading variants.
经过队列内分析以及队列间的 meta-analyses ,在166个基因座中鉴定了244个独立变异。根据解剖结构,我们将244个全基因组显著变异分为10个面部区域(补充注释),包括前额、眉间、眼睛、颞下颌、颧骨、鼻子、上颌、上颚、下颚和下颌。鼻子是十个区域中与大多数变体(107个)相关的特征。计算了每个3D面部片段在每个水平上的全基因组遗传力。3D面部节段的遗传率在7.47%至52.3%之间。正如我们所预期的,鼻节段也是最具遗传性的区域之一(42.17%至46.86%)。 在全基因组阈值下的244个主要变异中,有130个是新的,而在全研究阈值下的151个变异中,65个是新变异。当一个基因座与先前报道的与面部变异相关的基因组基因座不重叠时,我们认为它是新的。因此,166个基因座中有62个是新的。通过PhenoScanner,一个基于网络的GWAS库(方法)研究了244个变体与其他复杂性状的相关性(P<1×10−5)。我们发现,与面部形状共享高遗传成分的性状主要涉及身体测量、身体成分和头发形态。最高的相关性是心房颤动的风险,这表明面部特征可能是心血管疾病的生物标志物。

Characteristics of specific variants in EAS and EUR.
将研究中发现的244个主要变异与最近发表的EUR研究中报告的203个主要变异进行比较,89个变异是公有的
从而定义了三个不同的组:89个共享变体,155个EAS特异性变体和114个EUR特异性变体。
群体特异性变体组在各自的群体中具有更高的MAF,两个群体之间的共同变异没有MAF差异(bcd)
不难发现种群之间的相关基因具有基本相似的生物学过程(fgh)
不同人群的面部变异可能归因于随机漂移和自然选择。群体特异性变异解释了更大比例的自然选择,而共享变异可能主要解释影响面部变异的随机漂移。

Polygenic shape analysis generalizes results from EAS to EUR.
(构建三组平均面部)我们通过将(PPSEUR−PPSEAS)/2分别加上和减去人口中性平均面部来构建EUR和EAS PPS衍生的面部,该面部被构建为EUR和EAS人群平均形状的平均值(方法)。我们使用EAS和EUR个体的3D面部扫描来计算每个群体的平均面部,从而生成EUR和EAS的平均面部。当我们将分化的累积遗传效应放大五次时,PPS衍生的人脸看起来与EAS和EUR的实际平均人脸非常相似
将使用所有244个前导变体的整个面部的EUR和EAS PPS衍生的面部与使用从基因组中随机选择的244个变体的整个脸部的PPS进行比较。证明与使用随机变异时相比,使用所有244个前导变体的整个面部的EUR和EAS PPS衍生的面部与真实的人群平均人脸更相似。
PPS在EUR和EAS之间的大多数面部区域构建了形态变异。研究结果表明,使用前向变体的PPS衍生人脸在视觉和统计上都与全球和局部范围内的真实人群平均形状相似,这表明EAS研究中确定的面部形状效应很好地适用于EUR人群。

Variants contribute to EAS-FA.
具有阳性EAS-FA贡献的变体可能导致EAS群体的面部形态增加EAS-FA。相反,具有负EAS-FA贡献的变体可能导致EAS人群增加EUR面部外观。通过过滤的13种变体被认为增加了EAS-FA
主要通过查阅资料分析各个变体对于面部特征的影响。
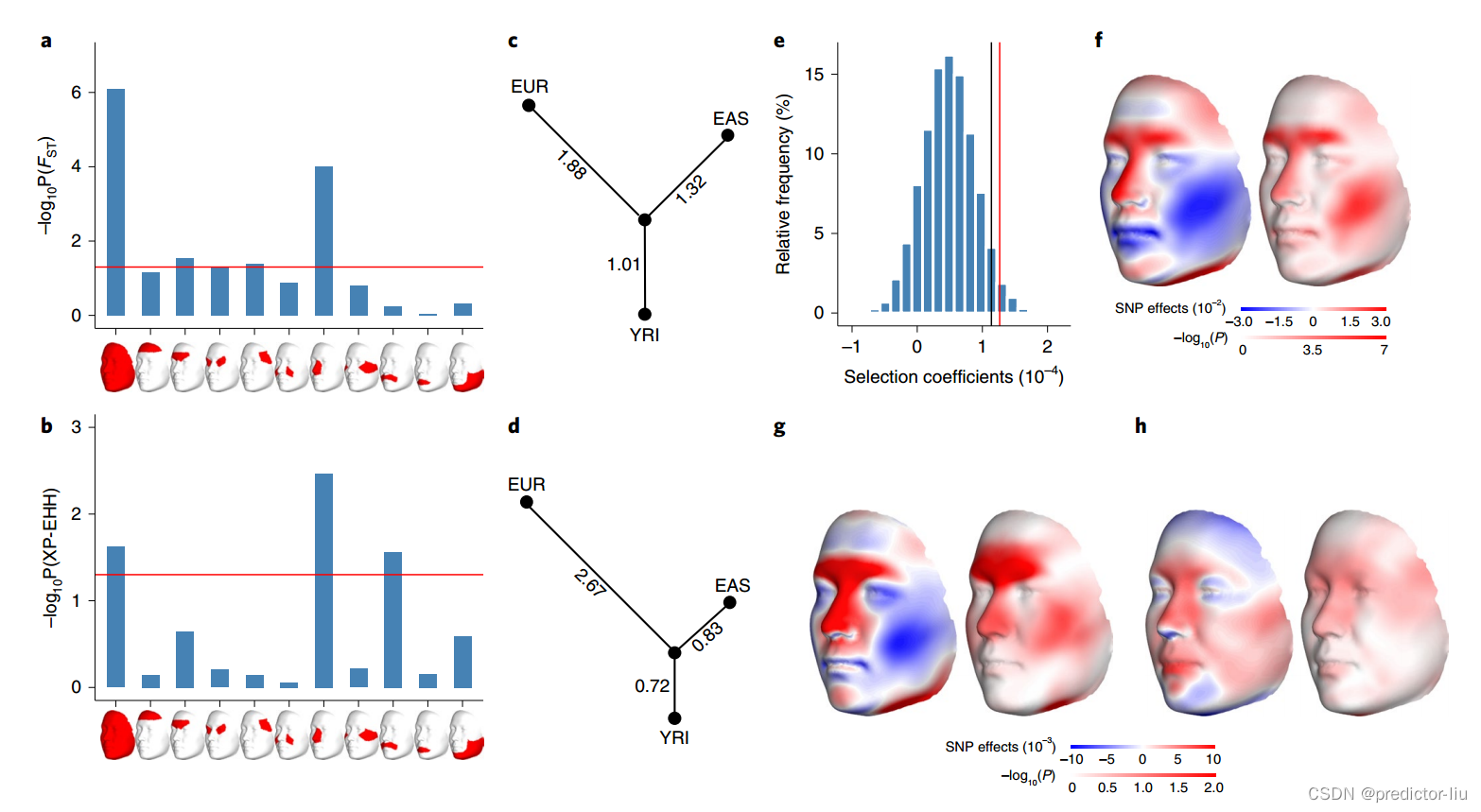
Nose-associated variants are under positive selection.
EAS和EUR人群中面部相关变异分化的自然选择分析和富集试验。为了评估EAS和EUR人群面部形态的变化是由于自然选择还是随机漂移,对发现的主要变异进行了几次选择分析。
FST富集测试和XP-EHH富集分析显示:整个面部和鼻子的区域的FST显著高于随机变体,表明面部形态在EAS和EUR中一直处于自然选择之下
推测:EUR–EAS差异背后的面部特征可能是由于EUR人群中发生的适应性选择,这使得欧洲血统的人群鼻子突出和狭窄,与东亚血统的人群明显不同
正文结束
---------------------------------------------------------------
本文涉及到的基因组学的知识以及研究方法和遗传学的评估方法仍然需要进一步探究