RNAseq常规做法是先筛选出差异表达基因,然后针对筛选出来的差异基因做GO富集分析。
1.水稻、小麦GO信息提取
在进行GO富集分析前首先要确定在你所使用的GO富集分析软件(包)中你所研究的物种有没有相应Gene与GO对应关系。如果没有,则需要自己根据该物种功能注释文件去提取Gene与GO对应关系。当然如果你对一些新组装出来的基因感兴趣,也可以通过跑blast2go等电子注释方法获取Gene对应GO信息。
## 方法一:从功能注释文件中提取。
##1.水稻(wget下载):wget https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_representative_annotation_2020-12-02.tsv.gz
setwd("/Users/mac/Desktop/项目/rice")
library(tidyr)
library(stringr)
#Note:提取注释文件中的GO-GENE
Func_Anno<-read.table("IRGSP-1.0_representative_annotation_2020-12-02.tsv",sep="\t",quote = "",header=TRUE,fill=TRUE)
GO_Gene<-Func_Anno[,c(2,10)]
#去掉空值行(基因对应GO id为空)
GO_Gene<-GO_Gene[-which(GO_Gene$GO==""),]
#拆分第二列数据(Note:一个逗号一个GO)
GO_Sep<-separate(GO_Gene,col=GO,sep =',',remove = TRUE,into=as.character(c(1:50)))
#检查GO是否完全分开
which(!is.na(GO_Sep[,51])) #判断最后一列是否有非NA值
sum(is.na(GO_Sep[,51])) #判断最后一列为NA值行数是否与矩阵行一样
#删除全为NA的列
GO_Sep<-GO_Sep[,-which(apply(GO_Sep,2,function(x) all(is.na(x))))]
#按行合并
GO_Gene_Matr<-data.frame(matrix(NA,300000,2))
for(i in 2:ncol(GO_Sep)){
if(i==2){
tmp<-as.matrix(GO_Sep[,c(1,i)])
GO_Gene_Matr<-tmp
}else{
tmp<-as.matrix(GO_Sep[,c(1,i)])
GO_Gene_Matr<-rbind(GO_Gene_Matr,tmp)
}
}
#取出含GO的行
GO_Gene_Matr<-GO_Gene_Matr[grep("GO:",GO_Gene_Matr[,2]),]
GO<-data.frame(str_sub(GO_Gene_Matr[,2],start=-11L,end=-2L))
GO_Gene_Matr_Final<-cbind(GO_Gene_Matr,GO)
GO_Gene_Matr_Final<-GO_Gene_Matr_Final[,c(1,3)]
GO_Gene_Matr_Final<-unique(GO_Gene_Matr_Final)
colnames(GO_Gene_Matr_Final)<-c("GeneID","GO")
write.table(GO_Gene_Matr_Final,"GO_Gene.txt",col.names = TRUE,row.names = FALSE,quote=FALSE,sep="\t")
##2.小麦(wget下载):wget https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v1.0/iwgsc_refseqv1.0_FunctionalAnnotation_v1.zip
Note:解压后使用里面的iwgsc_refseqv1.0_FunctionalAnnotation_v1__HCgenes_v1.0.TAB(高可信任基因)
setwd("/Users/mac/Desktop/项目/triticum_aestivum/GO")
library(tidyr)
library(stringr)
#1.提取注释文件中的GO-GENE-Function
Func_Anno<-read.table("iwgsc_refseqv1.0_FunctionalAnnotation_v1__HCgenes_v1.0.TAB",sep="\t",quote = "",header=TRUE)
GO_Gene<-Func_Anno[,c(1,8)]
#去掉空值行(基因对应GO id为空)
GO_Gene<-GO_Gene[-which(GO_Gene$GO.IDs..Description..via.Interpro==""),]
GeneID<-sub('..$','',GO_Gene[,1])
GO_Gene$Gene.ID<-GeneID
#拆分第二列数据
GO_Sep<-separate(GO_Gene,col=GO.IDs..Description..via.Interpro,sep =';',remove = TRUE,into=as.character(c(1:20)))
#检查GO是否完全分开(没有增加前面的into范围)
which(!is.na(GO_Sep[,21])) #判断最后一列是否有非NA值
sum(is.na(GO_Sep[,21])) #判断最后一列为NA值行数是否与矩阵行一样
GO_Sep_Matr<-data.frame(matrix(NA,300000,2))
k=1
for(i in 1:nrow(GO_Sep)){
for(j in 2:ncol(GO_Sep)){
if(is.na(GO_Sep[i,j])==FALSE){
GO_Sep_Matr[k,1]<-GO_Sep[i,1]
GO_Sep_Matr[k,2]<-GO_Sep[i,j]
k=k+1
}
}
}
GO_Sep_Matr<-GO_Sep_Matr[-which(apply(GO_Sep_Matr,1,function(x) all(is.na(x)))),]
Extract<-str_split_fixed(GO_Sep_Matr[,2]," ",n=2)
GO_Info<-cbind(GO_Sep_Matr[,1],Extract[,1:2])
#去重复
GO_Info<-unique(GO_Info)
colnames(GO_Info)<-c("GeneID","GO","Function")
GO_Gene<-GO_Info[,1:2]
write.table(GO_Gene,"GO_Gene.txt",col.names = TRUE,row.names = FALSE,quote=FALSE,sep="\t")
write.table(GO_Info,"GO_Info.txt",col.names = TRUE,row.names = FALSE,quote=FALSE,sep="\t")
#方法二:从一些数据库中下载背景文件,然后分析。
http://systemsbiology.cau.edu.cn/agriGOv2/download.php
2.GO富集分析
Note:参考TBtools完成。
2.1富集分析
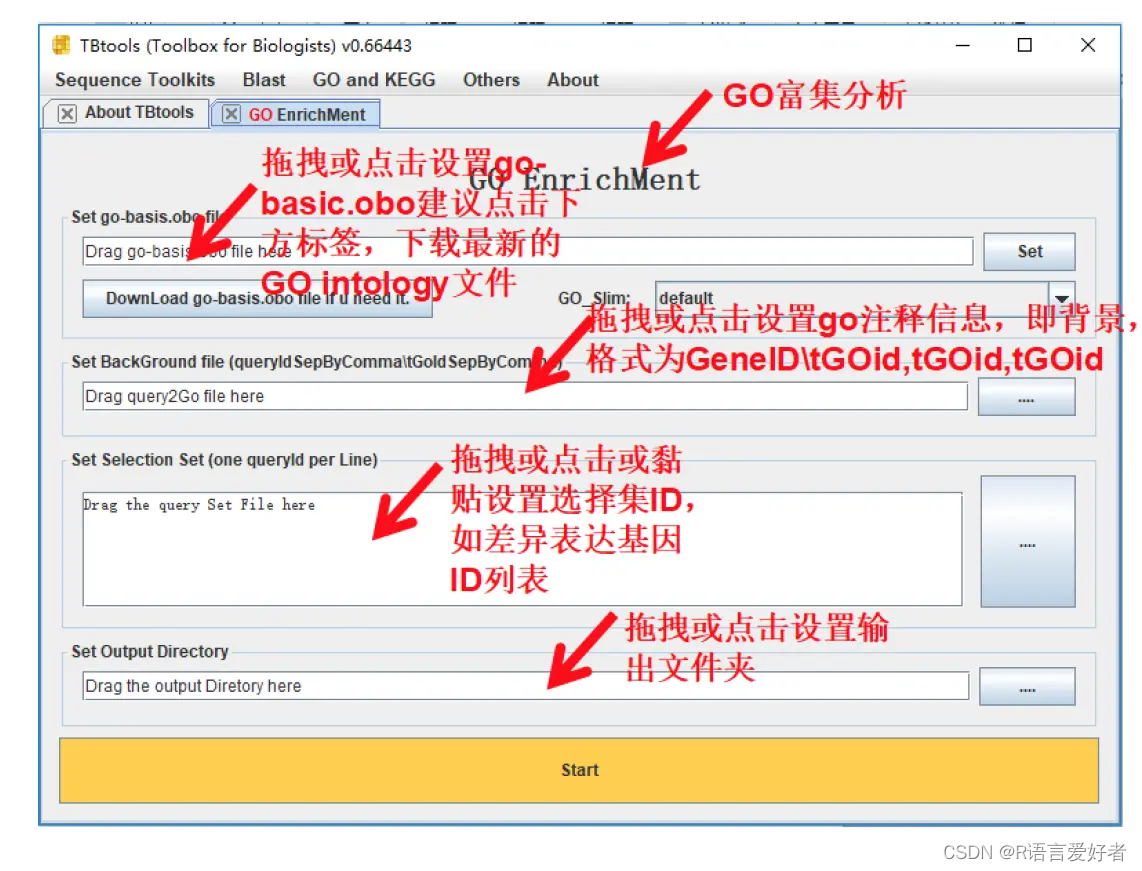
2.2可视化

转载链接:
https://www.jianshu.com/p/2cb4c83f8141