大家好,我是邓飞。前一段时间有小伙伴在星球提问:想将不同版本的SNP数据合并,不想重新call snp,想把绵羊的V2和V4版本的数据合并,具体来说,是V2转为V4然后与V4合并。

我建议用liftOver软件进行处理,并许诺写篇博客介绍一下。
还有小伙伴想把1.2的参考基因组,变为3.1的,问我如何处理,我还是建议用liftOver,在线网站也可以解决,但是本地编程更快一些。

1. 不同基因组转换对应关系原理
每一次参考基因组的更新,位置信息会有所变化,有些是插入了一些,有些是平移,有些是没有改变。
但是,每一个版本的参考基因组,都有对应的关系,如果我们根据对应的关系,就可以把旧版本的更新到新版本的位置。
应用领域:不同参考基因组call snp的vcf数据,可以通过这种方式转换为同一基因组版本,然后合并。有些芯片设计时是不同的基因组版本,也可以通过这种形式,进行转换,然后合并。
2. liftOver软件下载
网址:http://hgdownload.cse.ucsc.edu/admin/exe/
有苹果系统和Linux系统,这里以Linux系统为例进行介绍。
3. 查找物种的基因组版本
网址:https://hgdownload.soe.ucsc.edu/downloads.html
常见的物种都有:

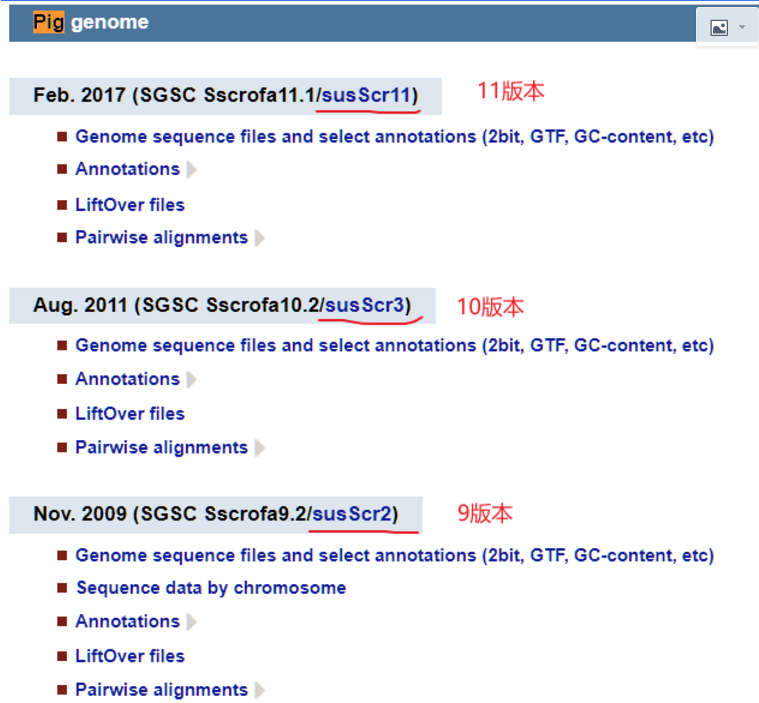
比如猪的版本有:
- V11
- V10
- V9
鸡的有:
- V6
- V5
- V4
牛的有:
- V9,V8,V7
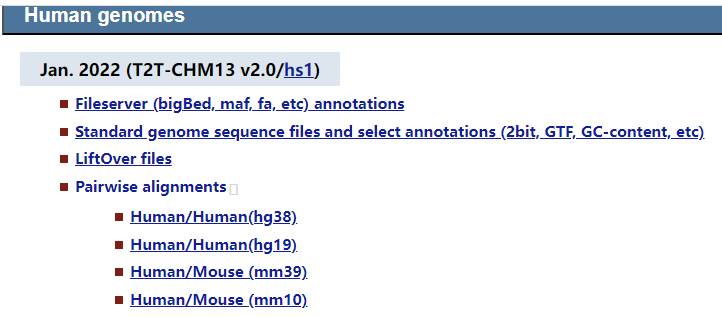
人的有:
- hg38
- hg19
- mm39
- mm10
4. 下载不同版本的liftOver数据文件
比如,这里以鸡为例子,进入网站:https://hgdownload.soe.ucsc.edu/goldenPath/galGal6/liftOver/
这里有V6变为V5,V6变为V4:,我们想把V6变为V5,可以下载:

当然,也可以V5变为V6,V4变为V6,只需要下载对应的chain文件即可:
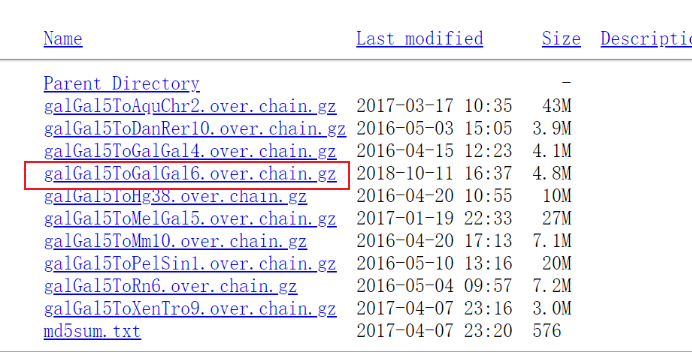
注意,下载的gz文件,不要解压缩。保持压缩状态
5. 整理位置信息
我们以plink数据为例,我们想把v5版的map变为v6版的map,首先将map数据变为bed的格式:
将位置信息整理为bed文件,可以根据map进行整理,染色体,开始位置,结束位置,没有行头。
只接受BED格式文件,BED格式文件只定义前三列:chr start end,无表头
注:end不等于start(如果是单位点的话,建议所有end = start+1)
转换代码:
sed 's/\s\+/ /g' new_v3.map >t1.map
awk '{print "chr"$1,$4,$4+1}' t1.map >tt.bed
6. 运行liftOver命令行转换
liftOver的语法为:
liftOver <输入文件> <chain文件> <输出文件> <unmapped文件>
示例代码:
将bed的V6版本,变为V5版本:
liftOver tt.bed galGal6ToGalGal5.over.chain.gz re_map.bed re_un_map.bed
- 第一个参数,tt.bed,就是bed文件,根据map生成的bed文件
- 第二个参数,是根据liftOver网站,下载的压缩文件,是对应关系,网址:https://hgdownload.soe.ucsc.edu/goldenPath/galGal5/liftOver/
- 第三个参数,是输出的结果文件
- 第四个参数,是没有匹配的结果文件
结果会输出成功转换的位点,和没有转换的位点。
为了方便我们后续使用,可以先运行一遍代码,将没有转换成功的位点删掉,然后再转换,这样就是一一对应的了。
有任何使用的问题,可以到关注公众号答疑。