论文
Microbiome differential abundance methods produce different results across 38 datasets
数据链接
https://figshare.com/articles/dataset/16S_rRNA_Microbiome_Datasets/14531724
代码链接
https://github.com/nearinj/Comparison_of_DA_microbiome_methods
这个人的github主页还有其他论文的数据和代码
https://github.com/jnmacdonald/differential-abundance-analysis 这个链接有很多冠以差异丰度分析的代码
这两天在看宏基因组的利用otu丰度数据做差异丰度分析,找到了这篇论文,看了摘要,好像是比较了不同差异丰度分析方法获得结果的异同。重复一下这里利用DESeq2做差异丰度分析的代码
这里我用到的数据集是
丰度数据
ArcticFireSoils_genus_table.tsv分组数据
ArcticFireSoils_meta.tsv
这里有一个疑问:论文提供的丰度表格数据有两个,还有一个是带rare后缀的,暂时不知道这两个有啥区别
首先是读取数据集
ASV_table <- read.table("metagenomics/dat01/ArcticFireSoils_genus_table.tsv",
sep="\t",
skip=1,
header=T,
row.names = 1,
comment.char = "",
quote="", check.names = F)
groupings <- read.table("metagenomics/dat01/ArcticFireSoils_meta.tsv",
sep="\t",
row.names = 1,
header=T,
comment.char = "",
quote="",
check.names = F)
dim(ASV_table)
dim(groupings)
groupings$Fire<-factor(groupings$Fire)这里需要对表示分组的赋予因子,要不然后面deseq2的步骤会有警告信息
判断一下丰度数据的样本名和分组数据的样本名顺序是否一致
identical(colnames(ASV_table), rownames(groupings))返回false不一致
对两个样本名取交集
rows_to_keep <- intersect(colnames(ASV_table), rownames(groupings))根据取交集的结果重新选择样本
groupings <- groupings[rows_to_keep,,drop=F]
ASV_table <- ASV_table[,rows_to_keep]疑问:这里中括号里的drop参数是啥作用了?
再次判断两个数据集中样本名的顺序
identical(colnames(ASV_table), rownames(groupings))这次返回TRUE
对分组文件的列名进行修改
colnames(groupings)[1] <- "Groupings"差异丰度分析
library(DESeq2)
dds <- DESeq2::DESeqDataSetFromMatrix(countData = ASV_table,
colData=groupings,
design = ~ Groupings)
dds_res <- DESeq2::DESeq(dds, sfType = "poscounts")
res <- results(dds_res,
tidy=T,
format="DataFrame",
contrast = c("Groupings","Fire","Control"))
head(res)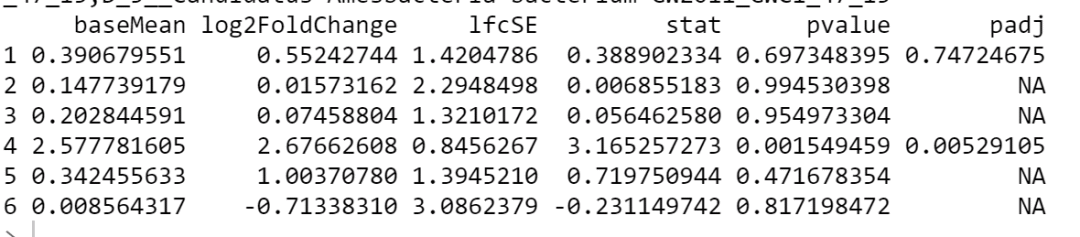
火山图代码
DEG<-res
logFC_cutoff<-2
DEG$change<-as.factor(ifelse(DEG$pvalue<0.05&abs(DEG$log2FoldChange)>logFC_cutoff,
ifelse(DEG$log2FoldChange>logFC_cutoff,"UP","DOWN"),
"NOT"))
this_title <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG[DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$change =='DOWN',]))
DEG<-na.omit(DEG)
library(ggplot2)
ggplot(data=DEG,aes(x=log2FoldChange,
y=-log10(pvalue),
color=change))+
geom_point(alpha=0.8,size=3)+
labs(x="log2 fold change")+ ylab("-log10 pvalue")+
ggtitle(this_title)+theme_bw(base_size = 20)+
theme(plot.title = element_text(size=15,hjust=0.5),)+
scale_color_manual(values=c('#a121f0','#bebebe','#ffad21')) -> p1
p1+xlim(NA,10)+ylim(NA,30) -> p2
library(patchwork)
p1+p2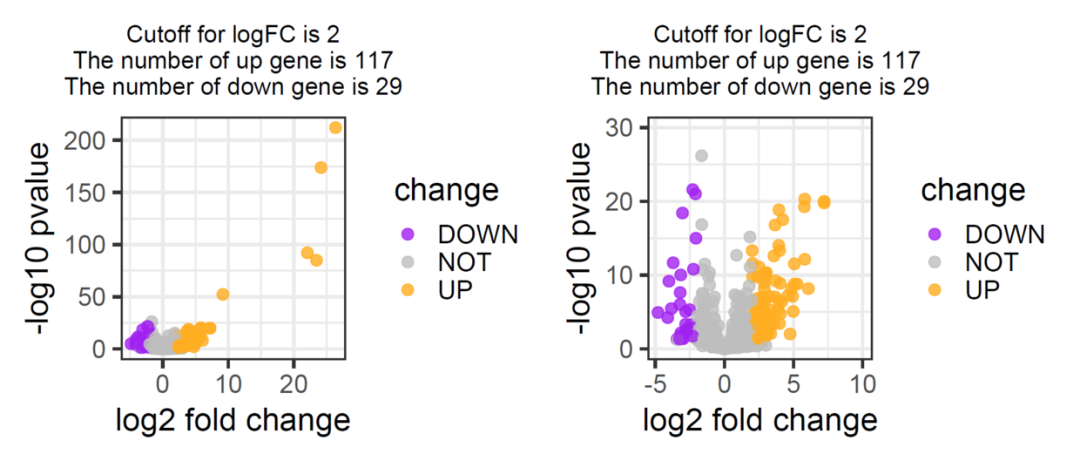
今天推文的示例数据和代码可以在昨天广告推文的留言区获取,点击蓝色文字直达昨天广告推文
欢迎大家关注我的公众号
小明的数据分析笔记本
小明的数据分析笔记本 公众号 主要分享:1、R语言和python做数据分析和数据可视化的简单小例子;2、园艺植物相关转录组学、基因组学、群体遗传学文献阅读笔记;3、生物信息学入门学习资料及自己的学习笔记!