摘要
微生物网络作为当下一种流行的数据分析方法被广泛应用于微生物群落研究。虽然目前已有许多并不断有新的微生物网络构建方法被开发出来,但与数据预处理、混杂因素、网络评估和解释相关的多个问题仍未得到足够的重视。因此,本文的目的是呼吁研究者们关注微生物网络构建和分析中这些悬而未决问题。

编译:鞠志成
英文标题:Open challenges for microbial network construction and analysis
中文标题:微生物网络构建与分析面临的挑战
期刊:ISME 2021
第一作者&通讯作者:Karoline Faust
作者单位:比利时鲁汶大学雷加医学研究所
引言
爱因斯坦曾说,提出一个问题往往比解决一个问题更重要。因为解决问题可能只是一个数学上或实验上的技能,而提出新的问题、新的可能性,从新的角度去看待旧的问题,却需要有创造性的想象力,这可能标志着科学的进步。本文的作者在此对微生物网络构建与分析提出十个开放式问题以供探讨,其中包括:
1. 不同类群间的相互作用是否影响微生物群落的组成?
2. 丰度数据应如何预处理?
3. 稀有类群如何处理?
4. 环境因素如何处理?
5. 高阶相互作用(HOI)如何处理?
6. 如何在计算机上评估微生物网络的构建?
7. 如何在生物数据上对微生物网络的构建进行标准测试?
8. 我们能从“一团乱麻”中知道什么?
9. 如何识别核心网络?
10. 微生物网络能在多大程度上地代表生态系统?
解决这些挑战将使微生物生态学家能够更好地将代表生物相互作用的边与其他边区分,这将反过来提高相互作用发现的实验验证的成功率。此外,用于微生物网络聚类和数据整合的工具将使其更容易识别具有生物学意义的分类群。最后,对网络和生态系统特性之间的联系的更好理解将使网络属性从单纯的数字变成有用的信息。总之,如果我们想从微生物网络中了解更多的信息,我们需要把研究重点扩大到推断算法之外,并解决这些挑战。
十大挑战
1.不同类群间的相互作用是否影响微生物群落的组成?
网络中的边通常被解释为微生物间的相互作用(如副产物的交叉喂养或营养物质的竞争),以这种方式预测的几种相互作用已被实验证实。然而,当相互作用可能太弱或不存在而无法影响群落组成,或是其采样空间或时间尺度的分辨率不足以检测到生态相互作用时,基于网络的推测仍然可以提供关于环境因素影响群落组成的见解,但如果群落动态在所选择的采样尺度上完全被随机过程主导,网络就不再能够提供有效信息,也就是说,网络构建的正确结果应该是一个“空网络”。因此,进行微生物群落动态的随机性(包括中性)和决定性分析测试,可以避免在无效的网络构建和误导性解释上浪费时间。尽管目前已经提出了一些测试方法,但它们很少在已知动态规则的真实的群落中得到验证。因此,第一个问题是开发和评估相互作用驱动的微生物群落动态测试,并将其应用于微生物网络推断中。
2.丰度数据应如何预处理?
由于提取、扩增和测序效率的差异,不同的样品中的总读数也不同,但这与细胞密度无关,因此不包含生物学信息。由于物种丰度与总读数呈共轭关系,因此通过某种形式的预处理是必要的,以防止虚假的网络关联。其中一种方法是稀疏化(即序列重抽),其本质上是随机地从样本中挑选读数,直到选定一个预较低的读数使每个样本读数保持一致。由于从一个特定分类群中选择读数的概率是由其在样本中的比例决定的,所以原始的分类群比例被保留下来,其不足之处在于它抛弃了一部分有效的数据,降低了微生物组的比较能力。然而,鉴于16S rRNA基因测序的高变异性,这一论点并不具有很大的份量。事实上,多次稀疏运算甚至被用来测试网络推断结果的稳健性。除了稀疏化之外,还有许多其他的预处理方法可用(其中最简单的是将读数转换为相对丰度),但在网络构建的背景下,关于它们性能的数据仍然非常少。通过实验确定的细胞密度也可用于调整总读数。这对网络构建是否有用取决于细胞密度的变化是由于生物相互作用,还是由于不具有生物学意义的外部因素。例如,如果营养物浓度的变化改变了细胞密度,但没有改变物种比例,那么改变细胞总数(单位体积)是一个需要消除的混杂因素。总之,第二个问题是比较不同的预处理方法在网络推断工具中的表现,以确定哪些组合效果最佳。
3.如何处理稀有类群?
测序数据中的大多数类群只在极少数样本中出现。这意味着测序数据的很大一部分是由“0”组成的。在生态统计数据中,零值可能代表真实的不存在或低于检测水平的存在(即分类群存在,但其DNA没有进入计数表)。在大多数样本中,两个分类群的零值匹配会产生强关联性,但如果实际上它们只在检测水平下随机变化,这种关联就会产生误导。目前有两种过滤方法来解决这一问题,它们都引入了一个任意的阈值:方法一是删除那些在较少样本中出现的类群(prevalence filter),方法二是当匹配的零值过大时,禁止计算成对类群之间的关联(Fig 1)。值得注意的是,当使用prevalence filter时,在进行下一步预处理前,应保留被丢弃分类群的总和,否则剩余分类群的相对丰度将被改变。无论是应用于单一分类群还是分类群之间,稀有类群的阈值设置都必须取得谨慎的平衡:如果它过于严格则会忽略零点所携带的宝贵信息,即在某些样本中很低比例的分类群。相反,如果过于宽松则不能解决由过多的零值造成的误差。
一些相关性计算方法,如Bray-Curtis不相似性在计算中忽略匹配零值,但当非零值对太少时,这种关联就不可靠了。因此,对匹配零值具有鲁棒性的相关性计算方法并不能避免定义一个任意的阈值来处理零值的必要性。Cougoul等人最近提出了计算零值的公式,由于最小/大的相关性系数都在置信区间内,超过该值就不可能再进行有意义的相关性分析。这该方法给出了零值的上限,是朝着正确方向迈出的一步。根据研究目的,稀有类群的问题也可以通过在较高物种分类水平分析来规避,如在纲而不是属的分类水平上进行分析。
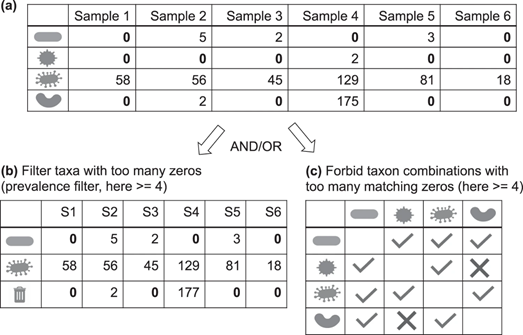
Fig 1 稀有类群过滤的两种方法
4.环境因素如何处理?
微生物群落的组成受到环境因素的强烈影响,如pH值、湿度、含氧量和营养物质。这些因素在大多数生态系统中的样品中不同,而微生物会对响应这种差异。因此,很难确定微生物网络中的一个边是由于对环境因素(或第三类群)的共同反应,还是代表两个类群之间的直接互动。有几种方法可以处理环境影响(Fig 2)。最简单的是将环境因素作为附加节点,并计算其与微生物类群的关联(Fig 2c)。通过CoNet和FlashWeave等工具中展示环境是如何构建微生物群落组成的。另一种策略是通过样本聚类或根据关键变量(如水深或健康状况)将样本分组,并分别为其建立网络(Fig 2d)。由于组内环境更加均质,特定组的网络随着环境变化而具有较少的边缘。在极端情况下,一些类群的(不)存在完全是由环境因素造成的。在这些情况下,计算相关性时忽略匹配零值(如FlashWeave的HE模式)相当于将样本拆分为组。这表明,环境异质性问题与之前的稀有类群的问题密切相关;一个类群之所以稀有,可能只是因为它存在于样本但不足以代表环境。除了将环境作为节点和样本分组策略外,还有一种方法是把环境因素回归并推断出不受环境影响的剩余丰度类群的相关性(Fig 2e)。然而,许多物种对环境参数的响应是非线性的,也就是说它们有一个最佳范围,当参数变化超过这个范围时,它们的生长就会下降。虽然回归可以扩展到处理这种非线性,但这增加了过度拟合数据的风险。最后,环境引起的间接边可以在网络构建后进行过滤(Fig 2f),例如,通过删除每个完全连接的三联体节点中具有最低交互信息的边。
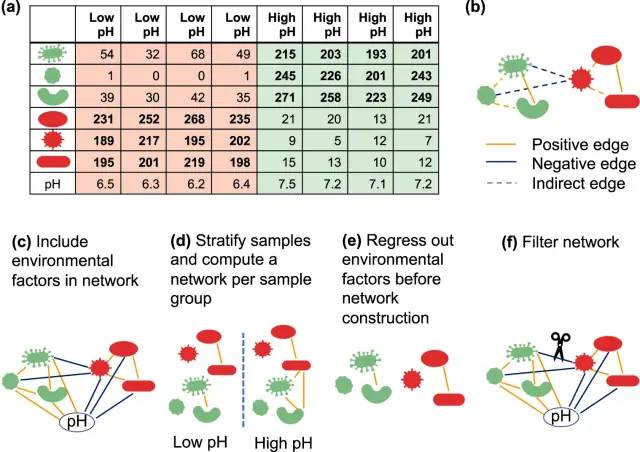
Fig 2 对环境异质性的处理方法
5.高阶相互作用(HOI)如何处理?
HOI的严格定义为受其他物种改变的多个物种之间的相互作用。例如,如果一种微生物依赖于另一种微生物分泌的物质,而第三种微生物也产生该物质,那么前两种微生物之间的交叉喂养关系就会被削弱。HOIs在模拟中影响群落的稳定性和多样性,并在实验中被证明可以改变宿主的生存能力。可以通过测量成对物种的生长曲线和在这些数据上对无HOI模型进行参数化来检测HOI,模型预测与群落行为的差异可能表明HOI的存在。然而,由于HOI-free模型可能由于HOI以外的其他原因而无法预测观察,因此该方法不能保证在狭义的修改交互作用中识别HOI。
大多数微生物网络构建工具忽略了HOI。此前,熵值最大化原则(寻找相关性使熵值函数最大化)已被用于从基因表达数据中推断基因间的HOIs。熵值最大化是否也能从微生物丰度数据中推断出HOIs,这是一个悬而未决的问题。在有/无数据的情况下,关联规则挖掘可以发现被解释为HOIs的逻辑规则。该规则的一个例子是物种A只在两个物种B和C的存在下才被发现(如它需要B和C分别生产的两个辅助因子),在此情况下,A和B或A和C之间的相互作用在第三个物种到来之前是不存在的,这可以看作是相互作用修改的一个极端情况。虽然以前报道过一些涉及两个以上微生物类群的关联规则,但不清楚这些是由于过度拟合(所有HOI推断算法都面临挑战)、环境因素、成对关联的组合还是真正的HOI。最后,HOI的可视化并非易事并需要超图(hypergraphs),即一条边连接两个以上节点的网络。解释和分析此类超图是HOIs的另一开放式挑战。
6.如何在计算机上评估微生物网络的构建?
进行评价是为了确定哪些工具能推断出最准确的网络,并探讨样本数量和其他数据属性如何影响工具的性能。鉴于缺乏全面的生物基准数据,微生物网络推断的评估在很大程度上仍然是在计算机上进行的。人工数据集是通过多种方法生成的,从用群体模型(通常是广义的Lotka-Volterra)进行模拟,到给定一个理想的相关矩阵产生多变量分布的统计方法(NorTA)。一般来说,工具性能对依赖于数据生成过程与工具假设的匹配程度很敏感。最好是数据生成过程是已知的,但对大多数微生物群落来说,并不能确切知道哪些过程塑造了物种丰度。因此,依赖于单一数据生成方法的评估将有利于那些假设恰好与数据生成背后的假设最接近的工具。为了避免这种偏差,计算机评估需要采用一系列包含不同噪声水平的数据模拟程序。另一个重点是将工具开发与评估分开,因为工具开发者很难客观地评估他们的工具。这些标准在评估框架中得到了实施,例如基因调控网络推断的DREAM问题。虽然微生物网络推断的相关问题仍然没有解决,但目前已经对微生物网络推断工具进行了一些独立的评估。有趣的是,在这两个评估(详见原文参考文献17,35)中,经典相关性方法如Pearson和Spearman的表现往往与更复杂的新网络推断算法一样好。这种令人惊讶的结果的一个可能的原因是,新算法在解决它们所设计的特定问题方面表现出色,例如在高度不均匀的物种组成数据中进行推断,而在其他同等重要的问题上表现较差,例如在有噪声的情况下进行推断。针对微生物网络的DREAM问题将为工具开发者提供更多异质性的基准数据,并最终提高工具在更多样化的环境中的表现。
7.如何在生物数据上对微生物网络的构建进行标准测试?
生物信息学工具基准测试的黄金标准是对已知结果的生物数据进行评估。对于微生物网络推断,这意味着在已知相互作用的群落的微生物测序数据上评估这些工具。尽管这样的数据集仍然很少,但仍然可以找到相关文献,例如一个已知的真核生物浮游植物相互作用的列表已被汇编并用于网络验证,大量的微生物相互作用在拟南芥根系群落中得到了实验验证。
然而,在对生物数据进行网络推断的基准测试时还存在几个问题。首先,不清楚已知的相互作用列表是否完整,因此,预测的相互作用是否是错误的还是根本没有观察到。因此,以前在自然界观察到的相互作用不能用来确定网络推断工具的准确性,而只能用于确定其敏感性,即工具发现已知相互作用的概率。这不足以作为工具比较的依据,因为一个工具可以通过简单地报告尽可能多的边来满足此标准。相反,当在受控条件下与小型群落合作时,所有的相互作用以及它们的缺失都可以被列举出来。然而,这时还不能确定这些相互作用在自然界中是否也是相关的。生物数据标准测试的第二个问题是,可能由于受到HOI的影响,推断的相互作用而不同于预期的相互作用,而不是推断本身的错误。解决此问题的一种方法是按照问题5中所述测试是否存在HOI。总之,相互作用已知且有测序数据的群落是构建微生物网络的基准的黄金标准。由于这样的数据集仍然很罕见,所以该问题是一个实验性的挑战:需要产生更多的这些基准数据以提高工具的性能。
8.我们能从“一团乱麻”中知道什么?
微生物网络推理算法通常会输出密集交互的“hairballs”,需要进一步分析以产生可测试的假设。但尽管有大量的推理工具,至今只有少数专门用于微生物网络的分析工具。两种类型的网络分析特别有参考价值:数据整合和聚类。网络特别适用于整合异质性数据。微生物的信息如特定基因的存在、环境偏好(如最适pH值)和已知的代谢能力可以映射到节点上,而已知的相互作用或其他推理工具的结果可以映射到边上。这些额外的数据有助于确认相互作用,并确定因对第三类群或环境因素的共同反应所产生的间接边。此外,外部数据的整合在某些情况下可能暗示了一种相互作用机制。例如一篇肠道细菌论文中报道的那样,如果A类群具有合成维生素B12途径而B类群缺乏B12合成的基因,它们之间的正向关系可能是由维生素B12交换介导的。
聚类将节点分配到不同组中,可以从头开始也可以使用预定义的聚类成员条件。第一种情况下网络聚类算法将趋向于与相同邻居相连的类群组合在一起,这样的类群通常会因环境因素(如pH值和温度)而发生共变,因此从头聚类是揭示生态位结构的一种手段,例如奶酪表面微生物网络中不同组的微生物类群对水分的响应不同。在第二种情况下,分类群根据已有知识分配到集群中,例如浮游生物功能类型或它们的系统。这简化了网络,而不再是了解数千个分类群之间的关系,而是十几个类群之间的关系。在这两种情况下,聚类还可以测试特定分类群或功能的富集程度,或与元数据相关联如与碳通量等。尽管它们对网络解释很有用,但执行这些任务的软件工具很少。因此,第八个挑战是开发更多处理数据整合和聚类分析的分析工具。
9.如何识别核心网络?
高通量测序使得对一个生态系统的许多实例进行测序,或对一个生态系统在不同时间点进行测序成为可能。同时,也出现了微生物网络是否跨空间或时间保存的问题。回答此问题的直接方法是为代表一个地区、条件或时间点的每个样本组分别构建一个微生物网络,然后计算这些网络的交叉点。生成的网络交集只包含所有特定网络中存在的边,因此可以解释为所关注生态系统的核心网络。核心网络的识别有几个问题,首先,只有当一个核心网络的边多于随机预期时才有意义,但选择哪种零模型来计算随机期望值并不清楚,这使核心网络的解释变得复杂。第二,边可能只在特定网络的一个子集中被保留下来。如果遇到此类边的频率比随机的频率更高,它们仍然是有意义的,然而它们会被全局交叉方法所遗漏。因此,核心网络的识别比简单地计算全局交集网络更具挑战性,需要专门的工具。此外,核心网络与Bashan等人讨论的通用网络并不完全相同,作者提出了一个测试交互网络普遍性的方法,这些网络在生态系统的不同实例中驱动群落动态。而核心网络是推断出来的网络,可能包含不代表相互作用的边。因此一个重要的核心网络的存在并不意味着群落动态是普遍的。
10.微生物网络能在多大程度上地代表生态系统?
微生物网络常被用于识别高度连接的节点,即所谓的枢纽(hub)。其理念是这些hubs是对生态系统具有特殊重要性的类群即keystones。这一观点有两个重要的假设:第一,枢纽类群可以被网络推断算法正确识别;第二,它们在生态系统中确实发挥着特殊作用。检验第一个假设的评估测试表明,网络推理算法并不总是能正确识别已知的枢纽节点,此外,很少有枢纽类群被实验证实是关键物种。因此,第二个假设的有效性目前是一个开放的问题。这引出了一个更普遍的问题,即网络是否能很好地代表生态系统,从而通过网络分析获得系统级的洞察力。假设网络推断足够准确,那么网络属性(如负相关边的百分比、模块化和网络密度等)能否提供关于所研究生态系统的有用信息?尽管一些理论研究已经探讨了网络属性对生态系统稳定性的影响,但实验证据仍然很少,而且并非与理论预期一致。因此,最后的挑战是要更深入地探索网络和生态系统特性之间的联系。
1. Faust K. Open challenges for microbial networkconstruction and analysis. ISME J. 2021 Nov;15(11):3111-3118..
原文链接:https://doi.org/10.1038/s41396-021-01027-4
中国科学院生态环境研究中心
环境生物技术重点实验室
邓晔 研究员课题组发布
编译:鞠志成