人类对于核酸的研究已经有100多年的历史。20世纪60年代末70年代初,人们致力于研究基因的体外分离技术。在此,pcr技术应运而生。
PCR(Polymerase Chain Reaction),即“聚合酶链式反应”,是为了解决在体外特异性地扩增某个基因的问题,从而满足对核酸的体外研究和应用。
pcr技术发展:
| 时间 | 大事记 |
| 1953 DNA结构基础 |
沃森和克里克发表的DNA双螺旋结构模型标志着分子生物学的诞生,这一成果也被誉为20世纪以来生物学方面最伟大的发现[1]。自此,人类一直在探索研究DNA的新方法,新技术。 |
| 1971 PCR技术雏形 |
Khorana第一个成功完成基因的合成—丙氨酸tRNA编码基因[2];Kleppe提出体外扩增设想[3]。但由于当时的技术水平有限,并且具有较强稳定性的DNA聚合酶还未被发现,DNA体外扩增技术并未获得全面推广。 |
| 1985 PCR技术首次提出 |
Mullis阐述了具有划时代意义的PCR技术[4]。其原理类似于DNA的体内复制,只是在试管中提供体外合成合适的条件---模版DNA,寡核苷酸引物,dNTP,DNA聚合酶,合适的缓冲体系,DNA变性、复性及延伸的温度与时间。最初使用的大肠杆菌DNA聚合酶Klenow片段不耐热,每次加热变性DNA后都要重新补加。耗时、费力、易出错。 |
| 1988 PCR技术的优化 |
Saiki等在黄石公园温泉中分离的一株水生嗜热杆菌(thermus aquaticus, Taq) 中提取到一种耐高温DNA聚合酶[5]。该酶耐高温的性质使其热变性时不会被钝化,不必在每次扩增反应后再加新酶,从而极大地提高了PCR扩增的效率。Taq酶的发现,使得PCR真正变为现实,为其自动化铺平了道路。 |
| 1989 PCR爆炸年 |
美国《Science》杂志列PCR为十余项重大科学发明之首,将热稳定DNA聚合酶命名为“年度分子”。 |
| 1996年 qpcr登上舞台 |
1996年,Real Time Quantitative PCR的发表标志着qPCR的诞生[4]。它引入了荧光标记的水解探针,并在每个PCR循环的延伸结束后采集荧光信号,将到达指数扩增期所需的循环数(Ct值,后经标准化统一为Cq值)与模板的初始浓度建立了关系 |
| 20世纪末 数字pcr出现 |
至20世纪末,Vogelstein 等提出数字PCR( digitalPCR,dPCR) 的概念,将一个样本分充分稀释,分配到不同的反应单元,每个单元包含少于或等于一个拷贝的目标分子( DNA 模板) ,在每个反应单元中进行单独、平行的PCR反应分,扩增结束后对各个反应单元的荧光信号进行统计学分析,以实现绝对定量及稀有等位基因的检测。 |
随着生物实验需求的不断发展,PCR技术在其发展的历程中,已经逐渐演化出一系列侧重于不同实验目的及应用的PCR分类,其中较为常见的包括:touchdown PCR、multiplex PCR、qPCR以ddPCR。
pcr试剂发展:
PCR试剂根据用途可分为:
1.提取试剂,包括了各种提取dna的常用试剂,和纯化所用的试剂
2.pcr扩增试剂:主要是taq酶,dntp(四种碱基),缓冲试剂(buffer),引物
3.产物检测试剂:琼脂糖,聚丙烯酰胺凝胶,aps,tmed,eb等等
而其中DNA聚合酶是pcr中比较重要的试剂:
在PCR技术中,DNA聚合酶起着至关重要的作用,从某种意义上说,DNA聚合酶的研究进展决定着PCR技术的发展趋势和应用范围,研究者们一直在努力探寻着酶学性能好、保真度高的PCR用DNA聚合酶。
| 时间 | 大事记 |
| 1985 PCR技术首次提出 |
Mullis阐述了具有划时代意义的PCR技术[4]。其原理类似于DNA的体内复制,只是在试管中提供体外合成合适的条件---模版DNA,寡核苷酸引物,dNTP,DNA聚合酶,合适的缓冲体系,DNA变性、复性及延伸的温度与时间。最初使用的大肠杆菌DNA聚合酶Klenow片段不耐热,每次加热变性DNA后都要重新补加。耗时、费力、易出错。 |
| 1985 Klenow酶 |
1985年美国PE-Cetus公司人类遗传研究室的Mullis等科学家发明了具有划时代意义的聚合酶链 式反应,但其最早使用的DNA聚合酶是⑶// DNA聚合酶I经胰蛋白酶或枯草杆菌蛋白酶部分水解生 成的C末端605个氨基酸残基片段,即Klenow酶。该片段保留了 DNA聚合酶I的5'—3'聚合酶和3'—5' 外切酶活性,但缺少完整酶的5'—3'外切酶活性。Klenow酶不耐高温,在60尤下就会很快变性失活,引物链延伸反应需在37T下进行,因此每次循环都要重新添加Klemovv酶 |
| 1988年 Taq DNA聚合酶 |
Saiki等在黄石公园温泉中分离的一株水生嗜热杆菌(thermus aquaticus, Taq) 中提取到一种耐高温DNA聚合酶[5]。该酶耐高温的性质使其热变性时不会被钝化,不必在每次扩增反应后再加新酶,从而极大地提高了PCR扩增的效率。Taq酶的发现,使得PCR真正变为现实,为其自动化铺平了道路。 |
| Tth DNA 聚合酶 | Tth DNA 聚合酶是是从嗜热性嗜热杆菌 HB8 中分离的一种分子量为 94 KD 的单亚基聚合酶,与 Taq酶相比在氨基酸序列上具有 88%的同源性,其基本特性与 Taq 酶相似,具有 5'→3' 聚合酶活性和 5'→3' 外切酶活性,不具 3'→5' 外切酶活性。最适温度在75℃,在 95℃时酶活性半衰期为 20 min。在高温条件下,当反应体系中存在 Mn2+,该酶具有很强的反转录酶活性,能有效地将 RNA 反转录成 c DNA。当鳌合了 Mn2+后再加入 Mg2+,则可以使该酶的聚合酶活性增 加,反转录产生的 c DNA 在此酶的作用下还可以进行 PCR 扩增,使得 c DNA 的合成与扩增用同一种酶。由于普通的逆转录酶如 M-MLV、ANV 等的最适反应温度为 37℃,在此温度条件下反应,RNA 分子形成的发夹、回环等高级结构很难克服,致使反转录合成过程效率大为下降,而 Tth DNA 聚合酶在较高的温度下进行逆转录,增加了引物杂交和延伸的特异性,减少了由于 RNA 二级结构引起的问题,所以现在被广泛用于反转录-聚合酶链式反应 (RT-PCR),对于从RNA 水平检测和分析基因表达十分有用。 |
| Vent DNA 聚合酶酶 |
Vent DNA 聚合酶是美国 New England Biolabs 公司由海底火山口生长的嗜热高温球菌中分离纯化 获得的,相对分子量为 85 KD。该酶具有强的 5'→ 3'DNA 聚合酶活性,兼有 3'→5' 外切酶活性,没有检 测到 5'→3' 外切酶活性。Vent DNA 聚合酶是被分离 得到的第一个具有 3′→5′核酸外切酶活性的热稳定 DNA 聚合酶。3'→5' 外切酶活性的主要功能是校对作 用,当加入的核苷酸与模板不互补而游离时则被 3'→ 5' 外切酶切除,以便重新在这个位置上聚合对应的核 苷酸,从而有效降低碱基错误掺入率,提高扩增结果 的忠实性。因此,Vent DNA 聚合酶是一种高保真的 耐热 DNA 聚合酶,其保真度比 Taq DNA 聚合酶高 5~15 倍。Vent DNA 聚合酶热稳定性极好,97.5℃下半衰期长达 130 min,且该酶扩增长片断(>12 Kb)的功能较强。 |
| Pfu DNA 聚合酶 |
Pfu DNA 聚合酶来自激烈热球菌。该酶分子量约为 90 KD,具有 5'→3'DNA 聚合酶和 3'→5' 外切酶活 随着分子生物学研究的不断发展和深入,PCR 技 |
Taq DNA聚合酶发现了几十年,不但没有被取代,反而在分子生物学领域具有越来越广泛的应用。这一方面依赖于它自身的功能和特点,另一方面也依赖于研究人员对其不断的进化与改造。通过定点突变、结构域重组、定向进化等技术,Taq DNA聚合酶获得了许多催化性能改善或展现新功能的突变体,拓宽了酶的应用范围,下面就来做一个回顾。
-
通过理性设计得到的突变体
①缺失突变体。Taq DNA聚合酶的第一个突变体就是N端删除大片段了,根据删除长度的差异,有不同的名称:KlenTaq(∆236)(Barnes, 1992),Klentaq1(∆280)(U.S. Patent 5436149),Stoffel(∆289)(Lawyer, 1993),它们本质上是一样的,都是5’-3’外切结构域缺失的突变体。N端删除Taq的耐热型、盐离子耐受能力、保真性都有所提高,但持续合成能力显著下降,对引物3’端错配的敏感性明显降低,这说明5’-3’外切结构域对酶的整体构象可能有很大的影响。研究人员发现大肠杆菌pol I后,又发现一种N端截短的Klenow片段,后者在DNA扩增上具有更好的表现,因此,研究人员得到Taq DNA聚合酶后,发现它与pol I有很多相似之处,自然而然的会进行这种尝试。根据结构或功能相似的酶对目标酶进行结构域编辑或者氨基酸替换,是一种常用的分子改造策略。
②Sanger测序突变体。使用Taq作为测序酶时发现,Taq无法掺入顺利ddNTP(除ddGTP),Tabor等(Tabor, 1995)通过F667Y突变体解决了这个问题。但是Taq(F667Y)掺入ddNTPs的速率不均匀,强烈偏向ddGTP,Li等(1999)在Klentaq1与ddNTP复合物晶体结构的基础上,对R660残基进行突变,结果R660D/F667Y双突变体掺入ddGTP与其它双脱氧核糖核苷酸的速率基本一致。根据酶-底物复合物的晶体结构对酶进行理性设计是一种常用的酶进化方式,当然也比较有难度,很多时候酶-底物的晶体甚至酶的晶体不易得,即使得到晶体结构,也很难判断关键的氨基酸。通常的做法是根据酶-底物的分子相互作用,扫描活性口袋的氨基酸,这种方法经常能在增强底物亲和力、拓宽底物谱方面得到满意的结果。 -
通过定向进化得到的突变体
定向进化是一种十分强大的酶分子改造技术,与理性设计的“所见即所得”不同,它的的特点是“所筛即所得”。定向进化允许构建庞大的突变体库,这就让酶有了各种可能(在自然界中这可能需要几百万年以上的时间),只要找到高效的筛选方法,一般都能得到令人惊喜的结果。构建突变体的方法有易错PCR、DNA改组、交错延伸PCR、体外随机延伸PCR等,筛选一般包括初筛与复筛,初筛依靠添加筛选压力让目标突变体留存下来,复筛将留存下来的克隆进行性能比较,从而选出最优秀的突变体。筛选的主要压力来自于初筛,必须具有高通量的方法,因为突变体库多样性可达108-109,一般采用与显色化学反应,酶促反应偶联产生便于观察的信号。
2001年,Ghadessy等报道了一种分区自我复制(Compartmentalized Self-replication,CSR)技术,该技术通过油水混合,形成一个个微小的油包水体系,每个体系就好像一个独立的隔离室,含有携带Taq DNA聚合酶突变体的大肠杆菌、dNTP、PCR buffer和Taq DNA聚合酶的引物,油包水体系在高温条件下仍具有很高的稳定性,通过预热变性,大肠杆菌释放出Taq DNA聚合酶,通过PCR循环,在筛选压力条件下,具有高活性的突变体以自身基因为模板进行PCR扩增,无义突变体被淘汰,反应结束纯化被富集的DNA产物,即得到了优秀的突变体基因。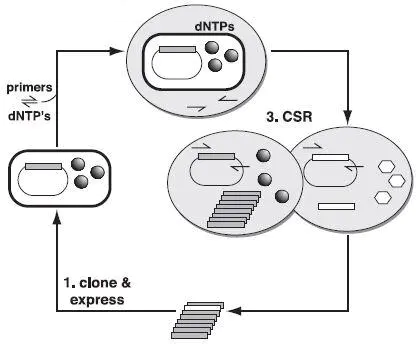
(Ghadessy F J, 2001)CSR总体方案。 (1)在大肠杆菌中克隆并表达聚合酶基因,球形代表有活性的聚合酶分子。 (2)将含有聚合酶和编码基因的细菌细胞悬浮在含有引物和dNTP的反应缓冲液中,并分离到水室中。 (3)聚合酶从细胞中释放出来,进行自我复制。 活性低的聚合酶(白色六边形)无法复制其编码基因。释放后代聚合酶基因并重新克隆,以进行另一轮CSR。
Ghadessy等得到两个突变体:T8(F73S, R205K, K219E, M236T, E434D, A608V),在97.5°C下的半衰期提高11倍,但催化效率和保真性有所下降;H15(K225E, E388V, K540R, D578G, N583S, M747R),对高浓度肝素抑制剂抗性提高130倍以上,但耐热性显著下降,催化活性也有下降。虽然本次研究没有得到非常优秀的突变体,但CSR技术为热稳定DNA聚合酶的定向进化提供了高效筛选方法,这种方法远比结果更有意义。
③热启动突变体。Kermekchiev等(Kermekchiev, 2003)以Klentaq为模板,通过易错PCR得到突变体库,然后利用一种基于放射性dNTP标记的在位筛选方法得到一个Klentaq Cs3AC(E626K,I707L)突变体,该突变体在37℃时活性明显降低,但68℃时仍保持正常活性,是一款极具潜力的热启动酶。2009年,该团队在Cs3AC的基础上对706-708残基进行饱和扫描突变,经过筛选得到一个突变体Klentaq 10(E626K, I707L,E708K),该突变体的全血抑制抗性、土壤抑制抗性、荧光染料抑制抗性比野生酶都有提高,研究人员将三个氨基酸替换平移到全长Taq DNA聚合酶上之后,得到一突变体Taq 22(E626K, I707L,E708Q)抑制剂抗性比野生Taq显著提高。Taq 22不但获取了冷敏感特征,提高了抑制剂抗性,还保持了野生Taq的热稳定性和催化活性,如果配合TP7抗体,具有“热启动”PCR试剂盒开发的潜力。
④逆转录活性突变体。野生型Taq DNA聚合酶没有逆转录活性,但Sauter等(Sauter, 2006)以KlenTaq为模板通过易错PCR和微孔板筛选,得到两个个逆转录活性提高的突变体:KlenTaq M1(L322M,L459M,S515R,I638F,S739G,E773G),KlenTaq M2(L322M,L459M,S515R,I638F,S739G,E773G,L789F)。为了将这种增强的逆转录活性用于基于探针的一步法RT-qPCR检测,Kranaster等(Kranaster, 2010)将KlenTaq M1的突变平移到全长Taq上,得到一个新突变体Taq M1,结果Taq M1即基本保留了野生Taq酶的PCR性能,又显著增强了逆转录活性。Blatter等(Blatter, 2013)进一步将M1的突变与M747K(Gloeckner, 2007)混组,得到一个新突变体RT-KTq2(L459M,S515R,I638F,M747K),RT-KTq2的逆转录能力比KlenTaq M1更强,并且DNA模板扩增能力也有所提高。RT-KTq2的高耐热性有利于提高RT-PCR的特异性,RT-KTq2无RNase H活性,降低了RNA损伤,因为兼具DNA聚合酶活性,可以实现了逆转-扩增一酶化。Blatter等用RT-KTq2对几个人源基因进行的基于SYBR的RT-qPCR检测,得到了不错的扩增曲线,但作者没有与商品化MMLV/Taq逆转录试剂盒对比,如果RT-KTq2能达到或接近MMLV的逆转录活性,那么RT-KTq2能够成为一款优秀的逆转录扩增酶。
⑤抑制剂抗性突变体。从各种各样的样本中进行核酸检测,是PCR的一个重要应用领域。一般的流程是,先从样本中将核酸提取出来,再用DNA聚合酶进行PCR扩增,最后根据核酸电泳条带或者荧光信号等方式观察扩增结果。有时候,因为样本的稀有性等原因无法提取DNA,或者基于检测时效性和便利性的考虑,我们希望能够直接从样本中扩增靶标基因,比如血液、动植物组织、土壤。这些样本中的复杂组分可能对DNA聚合酶有很强的抑制作用,失败的扩增会造成假阴性的误判,这促使研究人员通过分子改造提高聚合酶的抑制剂耐受性。野生型Taq DNA聚合酶对血液、土壤等抑制剂的耐受性很差,N端删除大片段的耐受性有所提高,但仍然难以满足要求。Kermekchiev等(Kermekchiev, 2009)通过定点饱和扫描突变得到的Klentaq 10和Taq 22突变体,可以耐受25%的全血和15%的土壤提取物,E708残基可能发挥了重要作用。相比一般的定向进化手段,CSR技术为获得高抑制剂耐受性突变体提供了基础。Baar等(Baar, 2011)选择了与Taq DNa聚合酶序列同源性较高的八个物种的聚合酶为亲本,通过StEP技术获得8T嵌合体文库,以泥炭提取物、焦油、腐植酸等抑制剂为筛选压力,经过三轮CSR,得到了一个优秀突变体2D9。2D9是由四种不同的聚合酶混组的嵌合体,在多种抑制剂存在条件下,耐受性都有显著的提高,并且保真度与野生Taq基本一致。 -
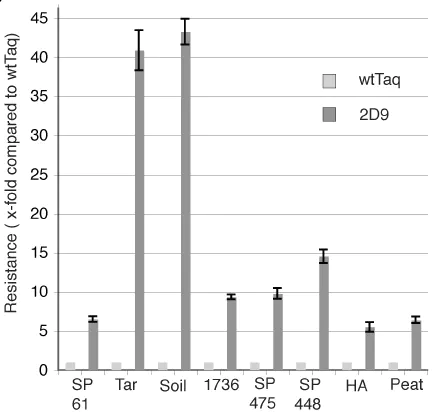
(Baar C, 2011)不同抑制剂下2D9对野生型Taq的相对活力
- 2014年,该团队(Arezi, 2014; Hogrefe, 2016)通过CSR得到一个新的全长Taq突变体Taq 2C2(G59W,V155I,L245M,L375V,E507K,E734G,F749I),对EDTA血耐受比例达到65%,对肝素血耐受达到25%,催化速率也比野生酶有所提高,可以作为直接血液核酸检测试剂盒的聚合酶。2016年,Schafer等以Taq-E507K为亲本,通过定向进化得到一个突变体Taq 15(A61T, K346E, S357C, E507K, I707M, F749I),该突变体对PCR buffer中的缓冲液、盐离子浓度具有极高的耐受性,可以很好的直接从葡萄、马铃薯叶片中扩增>1Kb的DNA片段,可用于植物叶片直扩PCR试剂盒的研发。
⑥快速延伸突变体。Taq DNA聚合酶在72℃时延伸速率大约是60nt/s,但实际中一般以1kb/min的速率扩增,一般一个PCR扩增下来,需要1-2h的时间。对于大样本量的检测或者时间要求急迫的检测,人们希望能够更快的扩增。Taq 2C2突变体的催化效率大约是野生Taq的2倍,氨基酸突变E507K对这种增强作用有重要意义。在一些报道中,直接提高Taq DNA聚合酶Kcat的突变体不多,但一般增加了模板亲和力或容错能力的突变体,往往在较短时间内扩增长片段的能力也同样增强。
⑦长片段扩增突变体。Taq DNA聚合酶在长片段扩增上,最卓越的成果是Klentaq1与Pfu混合酶(Barnes, 1994; U.S. Patent 5436149),可以扩增长达35kb的Lambda DNA片段,保真性也比Taq DNA聚合酶显著提高,但单独的Klentaq1持续合成能力是显著下降的(比Taq低10倍)。Ignatov等(US 11/658,610)将Tth DNA polymerase(该酶的扩增能力高于Taq,但区分对引物3’错配不敏感)的N端4-600多肽与Taq DNA polymerase的C端554-832多肽拼接成一个新的嵌合体酶Tth-Taq,嵌合体的扩增能力接近Tth,延伸能力比Taq酶显著提高。将具有DNA结合能力的ssDNA或dsDNA结合蛋白与Taq DNA聚合酶融合,也能提高酶的延伸能力,但真正意义上的长延伸突变体,还是Yamagami等(Yamagami, 2014)通过对E742和A743的扫描突变得到的。E742A和A743H两个突变体能够很好的从Lambda DNA上扩增15kb的长片段(条件:99℃ 5 s,66℃ 5 min,30个循环)。然而有趣的是,E742A/A743H双突变体的引物延伸能力比单突变强得多,但PCR效果却不如单突变体。凝胶迁移实验的结果表明双突变体与DNA产物有更强的结合能力(在电泳图上产生拖带),这可能不利于PCR起始阶段的扩增,导致PCR失败。我在融合Sso7d结构域与KOD DNA聚合酶时,也发现了类似的情况。有研究人员认为,Taq DNA聚合酶校正活性的缺失限制了其长片段扩增能力,因为随着目标产物长度的增加,延伸过程出错配的概率也就越高,阻挡了继续延伸,保真酶因为能切除错配,往往具有更强的持续延伸能力。这个解释符合事实,我认为长片段延伸突变体除了提高Taq DNA聚合酶的DNA亲和力,也可能增加了“容错”能力,实际上是降低了保真性,这类突变体的实际应用意义不太大,高保真聚合酶在这方面明显具更有优势。
⑧RNA聚合活性突变体。对Taq DNA聚合酶底物谱的改变,是其分子改造的一个重要成果之一,除了前面介绍的增强Taq DNA聚合酶掺入ddNTP、dNTP标记物等非标准脱氧核糖核苷酸的突变体,还有些研究将Taq DNA聚合酶变成了Taq RNA聚合酶。Xia等(Xia, 2002)以Stoffel片段为模板,通过基于噬菌体展示的定向进化,将其RNA聚合活性提高103-104。
Ong等(Ong J L, 2006)通过一种称为short-patch CSR的定向进化方法,得到一个突变体AA40(E602V,A608V,I614M,E615G)能够以与wt酶掺入dNTPs相同的催化效率掺入NTPs和dNTPs。
⑨跨损伤扩增突变体。Taq DNA聚合酶的跨损伤突变体,大多是由使用CSR技术而闻名的Holliger实验室完成的。d'Abbadie等(d'Abbadie, 2007)以Taq、Tth、Tfl三种聚合酶为亲本,经StEP混组和三轮CSR筛选得到若干突变体,扩增47,000-60,000年前的洞熊DNA时,成功率比野生酶显著提高。Loakes等(Loakes, 2009)通过CSR得到一个突变体5D4,能过聚合疏水性碱基类似物,后来Millar等(2015)将5D4用于重亚硫酸盐测序,结果发现5D4能够很好的跨过DNA损伤。此外,Obeid等(Obeid, 2011)以KlenTaq为模板,通过定向进化得到两个突变体I614K和M747K,它们获得了增强的跨损伤扩增能力。Obeid等I614K和M747K组合在一起后,双突变体的跨损伤扩增能力修复能力比单突变体显著增强。不过,这些突变体跨损伤能力的增强,是以牺牲保真性或者正常DNA扩增效率为代价的,它们应用范围比较狭窄,商业化价值不是太大。 -
通过结构域融合得到的突变体
⑩结构域融合突变体。一些来源于嗜热菌的的α-螺旋结构具有稳定DNA的作用,Pavlov等(Pavlov, 2002)将DNA拓扑异构酶V上一段重复的螺旋-发夹-螺旋结构融合到Taq DNA聚合酶的N端和C端,结果N端融合酶TopoTaq对Na+、K+、甜菜碱的耐受能力显著提高,并且耐热性也有很显著的增强。Davidson等(Davidson, 2003)将Taq DNA聚合酶的480-485残基替换为T3 DNA聚合酶的硫氧还蛋白结合域(TBD),结果融合体的合成能力提高了20-50倍,移码突变概率降低6-7倍。后来,Wang等(Wang, 2004)将来自Sulfolobus solfataricus的Sso7d双链DNA结合蛋白,融合在Taq和Stoffel片段的N端,结果发现S-Taq与S-Stoffel的持续合成能力比之前显著增强,融合蛋白与野生酶的Kcat基本一致,但Km比野生酶降低4-8倍,融合蛋白还增加了对盐离子的耐受性,令人兴奋的是融合蛋白还没有改变酶的耐热性和保真性。Sso7d的融合“红利”在家族B的Pfu DNA聚合酶上效果更明显,风靡市场的Phusion高保真聚合酶就是Pfu-sso7d。 -

(Wang Y, 2004)Taq,Stoffel和S-Stoffel的PCR效率比较。 以DNA(130 pg / ml)为模板,扩增子的大小显示在底部。 PCR程序为:95°C 20s; 94°C 5s,72°C 30s(A)或60s(B)或2min(C),20个循环; 72°C 7分钟。