通过xyz文件可视化小分子
导入库
from __future__ import print_function
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole
from IPython.display import SVG
from rdkit.Chem import rdDepictor
from rdkit.Chem.Draw import rdMolDraw2D定义mol转svg函数
def moltosvg(mol,molSize=(450,150),kekulize=True):
mc = Chem.Mol(mol.ToBinary())
if kekulize:
try:
Chem.Kekulize(mc)
except:
mc = Chem.Mol(mol.ToBinary())
if not mc.GetNumConformers():
rdDepictor.Compute2DCoords(mc)
drawer = rdMolDraw2D.MolDraw2DSVG(molSize[0],molSize[1])
drawer.DrawMolecule(mc)
drawer.FinishDrawing()
svg = drawer.GetDrawingText()
return svg
def render_svg(svg):
return SVG(svg.replace('svg:',''))jensengroup的xyz2mol,提供了将MIT许可下的xyz文件转换为RDKit mol实例的代码
"""
https://github.com/jensengroup/xyz2mol/blob/master/xyz2mol.py
"""
##
# Written by Jan H. Jensen based on this paper Yeonjoon Kim and Woo Youn Kim
# "Universal Structure Conversion Method for Organic Molecules: From Atomic Connectivity
# to Three-Dimensional Geometry" Bull. Korean Chem. Soc. 2015, Vol. 36, 1769-1777 DOI: 10.1002/bkcs.10334
#
from rdkit import Chem
from rdkit.Chem import AllChem
import itertools
from rdkit.Chem import rdmolops
from collections import defaultdict
import copy
import networkx as nx # uncomment if you don't want to use "quick"/install networkx
global __ATOM_LIST__
__ATOM_LIST__ = [x.strip() for x in ['h ', 'he', \
'li', 'be', 'b ', 'c ', 'n ', 'o ', 'f ', 'ne', \
'na', 'mg', 'al', 'si', 'p ', 's ', 'cl', 'ar', \
'k ', 'ca', 'sc', 'ti', 'v ', 'cr', 'mn', 'fe', 'co', 'ni', 'cu', \
'zn', 'ga', 'ge', 'as', 'se', 'br', 'kr', \
'rb', 'sr', 'y ', 'zr', 'nb', 'mo', 'tc', 'ru', 'rh', 'pd', 'ag', \
'cd', 'in', 'sn', 'sb', 'te', 'i ', 'xe', \
'cs', 'ba', 'la', 'ce', 'pr', 'nd', 'pm', 'sm', 'eu', 'gd', 'tb', 'dy', \
'ho', 'er', 'tm', 'yb', 'lu', 'hf', 'ta', 'w ', 're', 'os', 'ir', 'pt', \
'au', 'hg', 'tl', 'pb', 'bi', 'po', 'at', 'rn', \
'fr', 'ra', 'ac', 'th', 'pa', 'u ', 'np', 'pu']]
def get_atom(atom):
global __ATOM_LIST__
atom = atom.lower()
return __ATOM_LIST__.index(atom) + 1
def getUA(maxValence_list, valence_list):
UA = []
DU = []
for i, (maxValence, valence) in enumerate(zip(maxValence_list, valence_list)):
if maxValence - valence > 0:
UA.append(i)
DU.append(maxValence - valence)
return UA, DU
def get_BO(AC, UA, DU, valences, UA_pairs, quick):
BO = AC.copy()
DU_save = []
while DU_save != DU:
for i, j in UA_pairs:
BO[i, j] += 1
BO[j, i] += 1
BO_valence = list(BO.sum(axis=1))
DU_save = copy.copy(DU)
UA, DU = getUA(valences, BO_valence)
UA_pairs = get_UA_pairs(UA, AC, quick)[0]
return BO
def valences_not_too_large(BO, valences):
number_of_bonds_list = BO.sum(axis=1)
for valence, number_of_bonds in zip(valences, number_of_bonds_list):
if number_of_bonds > valence:
return False
return True
def BO_is_OK(BO, AC, charge, DU, atomic_valence_electrons, atomicNumList, charged_fragments):
Q = 0 # total charge
q_list = []
if charged_fragments:
BO_valences = list(BO.sum(axis=1))
for i, atom in enumerate(atomicNumList):
q = get_atomic_charge(atom, atomic_valence_electrons[atom], BO_valences[i])
Q += q
if atom == 6:
number_of_single_bonds_to_C = list(BO[i, :]).count(1)
if number_of_single_bonds_to_C == 2 and BO_valences[i] == 2:
Q += 1
q = 2
if number_of_single_bonds_to_C == 3 and Q + 1 < charge:
Q += 2
q = 1
if q != 0:
q_list.append(q)
if (BO - AC).sum() == sum(DU) and charge == Q and len(q_list) <= abs(charge):
return True
else:
return False
def get_atomic_charge(atom, atomic_valence_electrons, BO_valence):
if atom == 1:
charge = 1 - BO_valence
elif atom == 5:
charge = 3 - BO_valence
elif atom == 15 and BO_valence == 5:
charge = 0
elif atom == 16 and BO_valence == 6:
charge = 0
else:
charge = atomic_valence_electrons - 8 + BO_valence
return charge
def clean_charges(mol):
# this hack should not be needed any more but is kept just in case
#
rxn_smarts = ['[N+:1]=[*:2]-[C-:3]>>[N+0:1]-[*:2]=[C-0:3]',
'[N+:1]=[*:2]-[O-:3]>>[N+0:1]-[*:2]=[O-0:3]',
'[N+:1]=[*:2]-[*:3]=[*:4]-[O-:5]>>[N+0:1]-[*:2]=[*:3]-[*:4]=[O-0:5]',
'[#8:1]=[#6:2]([!-:6])[*:3]=[*:4][#6-:5]>>[*-:1][*:2]([*:6])=[*:3][*:4]=[*+0:5]',
'[O:1]=[c:2][c-:3]>>[*-:1][*:2][*+0:3]',
'[O:1]=[C:2][C-:3]>>[*-:1][*:2]=[*+0:3]']
fragments = Chem.GetMolFrags(mol, asMols=True, sanitizeFrags=False)
for i, fragment in enumerate(fragments):
for smarts in rxn_smarts:
patt = Chem.MolFromSmarts(smarts.split(">>")[0])
while fragment.HasSubstructMatch(patt):
rxn = AllChem.ReactionFromSmarts(smarts)
ps = rxn.RunReactants((fragment,))
fragment = ps[0][0]
if i == 0:
mol = fragment
else:
mol = Chem.CombineMols(mol, fragment)
return mol
def BO2mol(mol, BO_matrix, atomicNumList, atomic_valence_electrons, mol_charge, charged_fragments):
# based on code written by Paolo Toscani
l = len(BO_matrix)
l2 = len(atomicNumList)
BO_valences = list(BO_matrix.sum(axis=1))
if (l != l2):
raise RuntimeError('sizes of adjMat ({0:d}) and atomicNumList '
'{1:d} differ'.format(l, l2))
rwMol = Chem.RWMol(mol)
bondTypeDict = {
1: Chem.BondType.SINGLE,
2: Chem.BondType.DOUBLE,
3: Chem.BondType.TRIPLE
}
for i in range(l):
for j in range(i + 1, l):
bo = int(round(BO_matrix[i, j]))
if (bo == 0):
continue
bt = bondTypeDict.get(bo, Chem.BondType.SINGLE)
rwMol.AddBond(i, j, bt)
mol = rwMol.GetMol()
if charged_fragments:
mol = set_atomic_charges(mol, atomicNumList, atomic_valence_electrons, BO_valences, BO_matrix, mol_charge)
else:
mol = set_atomic_radicals(mol, atomicNumList, atomic_valence_electrons, BO_valences)
return mol
def set_atomic_charges(mol, atomicNumList, atomic_valence_electrons, BO_valences, BO_matrix, mol_charge):
q = 0
for i, atom in enumerate(atomicNumList):
a = mol.GetAtomWithIdx(i)
charge = get_atomic_charge(atom, atomic_valence_electrons[atom], BO_valences[i])
q += charge
if atom == 6:
number_of_single_bonds_to_C = list(BO_matrix[i, :]).count(1)
if number_of_single_bonds_to_C == 2 and BO_valences[i] == 2:
q += 1
charge = 0
if number_of_single_bonds_to_C == 3 and q + 1 < mol_charge:
q += 2
charge = 1
if (abs(charge) > 0):
a.SetFormalCharge(int(charge))
# shouldn't be needed anymore bit is kept just in case
# mol = clean_charges(mol)
return mol
def set_atomic_radicals(mol, atomicNumList, atomic_valence_electrons, BO_valences):
# The number of radical electrons = absolute atomic charge
for i, atom in enumerate(atomicNumList):
a = mol.GetAtomWithIdx(i)
charge = get_atomic_charge(atom, atomic_valence_electrons[atom], BO_valences[i])
if (abs(charge) > 0):
a.SetNumRadicalElectrons(abs(int(charge)))
return mol
def get_bonds(UA, AC):
bonds = []
for k, i in enumerate(UA):
for j in UA[k + 1:]:
if AC[i, j] == 1:
bonds.append(tuple(sorted([i, j])))
return bonds
def get_UA_pairs(UA, AC, quick):
bonds = get_bonds(UA, AC)
if len(bonds) == 0:
return [()]
if quick:
G = nx.Graph()
G.add_edges_from(bonds)
UA_pairs = [list(nx.max_weight_matching(G))]
return UA_pairs
max_atoms_in_combo = 0
UA_pairs = [()]
for combo in list(itertools.combinations(bonds, int(len(UA) / 2))):
flat_list = [item for sublist in combo for item in sublist]
atoms_in_combo = len(set(flat_list))
if atoms_in_combo > max_atoms_in_combo:
max_atoms_in_combo = atoms_in_combo
UA_pairs = [combo]
# if quick and max_atoms_in_combo == 2*int(len(UA)/2):
# return UA_pairs
elif atoms_in_combo == max_atoms_in_combo:
UA_pairs.append(combo)
return UA_pairs
def AC2BO(AC, atomicNumList, charge, charged_fragments, quick):
# TODO
atomic_valence = defaultdict(list)
atomic_valence[1] = [1]
atomic_valence[6] = [4]
atomic_valence[7] = [4, 3]
atomic_valence[8] = [2, 1]
atomic_valence[9] = [1]
atomic_valence[14] = [4]
atomic_valence[15] = [5, 4, 3]
atomic_valence[16] = [6, 4, 2]
atomic_valence[17] = [1]
atomic_valence[32] = [4]
atomic_valence[35] = [1]
atomic_valence[53] = [1]
atomic_valence_electrons = {}
atomic_valence_electrons[1] = 1
atomic_valence_electrons[6] = 4
atomic_valence_electrons[7] = 5
atomic_valence_electrons[8] = 6
atomic_valence_electrons[9] = 7
atomic_valence_electrons[14] = 4
atomic_valence_electrons[15] = 5
atomic_valence_electrons[16] = 6
atomic_valence_electrons[17] = 7
atomic_valence_electrons[32] = 4
atomic_valence_electrons[35] = 7
atomic_valence_electrons[53] = 7
# make a list of valences, e.g. for CO: [[4],[2,1]]
valences_list_of_lists = []
for atomicNum in atomicNumList:
valences_list_of_lists.append(atomic_valence[atomicNum])
# convert [[4],[2,1]] to [[4,2],[4,1]]
valences_list = list(itertools.product(*valences_list_of_lists))
best_BO = AC.copy()
# implemenation of algorithm shown in Figure 2
# UA: unsaturated atoms
# DU: degree of unsaturation (u matrix in Figure)
# best_BO: Bcurr in Figure
#
for valences in valences_list:
AC_valence = list(AC.sum(axis=1))
UA, DU_from_AC = getUA(valences, AC_valence)
if len(UA) == 0 and BO_is_OK(AC, AC, charge, DU_from_AC, atomic_valence_electrons, atomicNumList,
charged_fragments):
return AC, atomic_valence_electrons
UA_pairs_list = get_UA_pairs(UA, AC, quick)
for UA_pairs in UA_pairs_list:
BO = get_BO(AC, UA, DU_from_AC, valences, UA_pairs, quick)
if BO_is_OK(BO, AC, charge, DU_from_AC, atomic_valence_electrons, atomicNumList, charged_fragments):
return BO, atomic_valence_electrons
elif BO.sum() >= best_BO.sum() and valences_not_too_large(BO, valences):
best_BO = BO.copy()
return best_BO, atomic_valence_electrons
def AC2mol(mol, AC, atomicNumList, charge, charged_fragments, quick):
# convert AC matrix to bond order (BO) matrix
BO, atomic_valence_electrons = AC2BO(AC, atomicNumList, charge, charged_fragments, quick)
# add BO connectivity and charge info to mol object
mol = BO2mol(mol, BO, atomicNumList, atomic_valence_electrons, charge, charged_fragments)
return mol
def get_proto_mol(atomicNumList):
mol = Chem.MolFromSmarts("[#" + str(atomicNumList[0]) + "]")
rwMol = Chem.RWMol(mol)
for i in range(1, len(atomicNumList)):
a = Chem.Atom(atomicNumList[i])
rwMol.AddAtom(a)
mol = rwMol.GetMol()
return mol
def get_atomicNumList(atomic_symbols):
atomicNumList = []
for symbol in atomic_symbols:
atomicNumList.append(get_atom(symbol))
return atomicNumList
def read_xyz_file(filename):
atomic_symbols = []
xyz_coordinates = []
with open(filename, "r") as file:
for line_number, line in enumerate(file):
if line_number == 0:
num_atoms = int(line)
elif line_number == 1:
if "charge=" in line:
charge = int(line.split("=")[1])
else:
charge = 0
else:
atomic_symbol, x, y, z = line.split()
atomic_symbols.append(atomic_symbol)
xyz_coordinates.append([float(x), float(y), float(z)])
atomicNumList = get_atomicNumList(atomic_symbols)
return atomicNumList, charge, xyz_coordinates
def xyz2AC(atomicNumList, xyz):
import numpy as np
mol = get_proto_mol(atomicNumList)
conf = Chem.Conformer(mol.GetNumAtoms())
for i in range(mol.GetNumAtoms()):
conf.SetAtomPosition(i, (xyz[i][0], xyz[i][1], xyz[i][2]))
mol.AddConformer(conf)
dMat = Chem.Get3DDistanceMatrix(mol)
pt = Chem.GetPeriodicTable()
num_atoms = len(atomicNumList)
AC = np.zeros((num_atoms, num_atoms)).astype(int)
for i in range(num_atoms):
a_i = mol.GetAtomWithIdx(i)
Rcov_i = pt.GetRcovalent(a_i.GetAtomicNum()) * 1.30
for j in range(i + 1, num_atoms):
a_j = mol.GetAtomWithIdx(j)
Rcov_j = pt.GetRcovalent(a_j.GetAtomicNum()) * 1.30
if dMat[i, j] <= Rcov_i + Rcov_j:
AC[i, j] = 1
AC[j, i] = 1
return AC, mol
def chiral_stereo_check(mol):
Chem.SanitizeMol(mol)
Chem.DetectBondStereochemistry(mol, -1)
Chem.AssignStereochemistry(mol, flagPossibleStereoCenters=True, force=True)
Chem.AssignAtomChiralTagsFromStructure(mol, -1)
return mol
def xyz2mol(atomicNumList, charge, xyz_coordinates, charged_fragments, quick):
# Get atom connectivity (AC) matrix, list of atomic numbers, molecular charge,
# and mol object with no connectivity information
AC, mol = xyz2AC(atomicNumList, xyz_coordinates)
# Convert AC to bond order matrix and add connectivity and charge info to mol object
new_mol = AC2mol(mol, AC, atomicNumList, charge, charged_fragments, quick)
# Check for stereocenters and chiral centers
new_mol = chiral_stereo_check(new_mol)
return new_mol定义xyz转mol对象函数
def mol_from_xyz(filepath):
charged_fragments = True # alternatively radicals are made
# quick is faster for large systems but requires networkx
# if you don't want to install networkx set quick=False and
# uncomment 'import networkx as nx' at the top of the file
quick = True
atomicNumList, charge, xyz_coordinates = read_xyz_file(filepath)
mol = xyz2mol(atomicNumList, charge, xyz_coordinates, charged_fragments, quick)
# Canonical hack
smiles = Chem.MolToSmiles(mol, isomericSmiles=True)
m = Chem.MolFromSmiles(smiles)
return m获取文件列表
from glob import glob
xyz_filepath_list = list(glob('./structures/*.xyz'))
xyz_filepath_list.sort()
print('total xyz filepath # ', len(xyz_filepath_list))total xyz filepath # 5197可视化分子
# Visualize molecules.
for i in range(5):
filepath = xyz_filepath_list[i]
mol = mol_from_xyz(filepath)
smiles = Chem.MolToSmiles(mol, isomericSmiles=True)
print(f'{i}-th file {filepath}: SMILES {smiles}')
display(render_svg(moltosvg(mol)))
num_mols = 20
mol_list = []
for i in range(num_mols):
filepath = xyz_filepath_list[i]
mol = mol_from_xyz(filepath)
if mol is None:
print(f'{i}-th file {filepath} failed, skip')
mol_list.append(mol)from rdkit.Chem import Draw
img = Draw.MolsToGridImage(mol_list, molsPerRow=5, subImgSize=(250, 250), maxMols=num_mols,
legends=None, useSVG=True)
img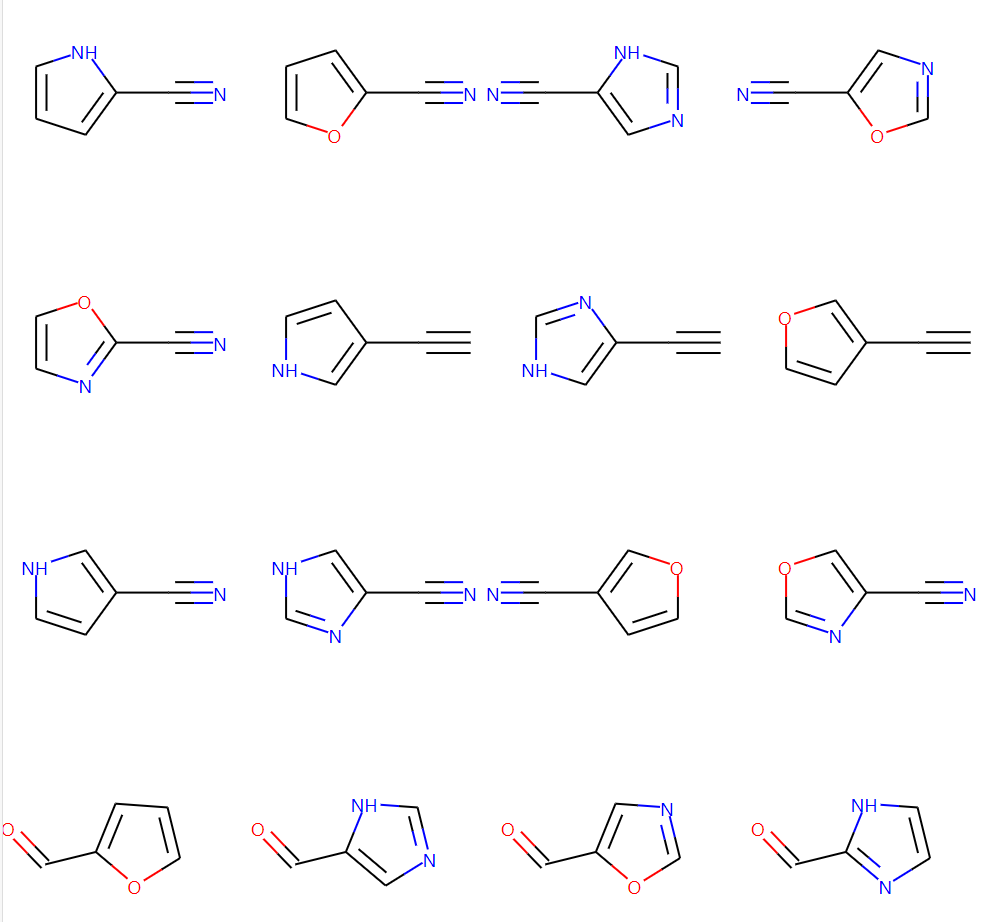
参考:
[1] https://github.com/jensengroup/xyz2mol/blob/master/xyz2mol.py
