摘要
本文开发了虚拟chip-seq,整合了基因表达和结合的关联信息,使用了来自其他细胞类型的TF结合位点,以及新细胞类型中染色质可及性数据,能够预测新细胞类型中单个TF的结合。该方法优于仅基于序列偏好预测TF结合的方法,预测了36个TF的结合(MCC > 0.3)。
解决的问题
DNA的一级结构(序列)、二级结构(形状)和三级结构(构象)都在TF结合中发挥作用,许多TF是间接和DNA结合的。在此情况下,使用体外数据训练的模型在进行体内实验时表现不佳。为了解决该问题,本文从探索依赖于上下文的TF bunding出发来解决这一问题。
评价指标
使用ENCODE提出的评估TF结合预测的指南:评估FN预测的受试者操作特征曲线(auROC)下的面积和评估FPs的精确回忆曲线(auPR)下的面积。
使用马修斯相关系数(MCC)评估模型在预定义阈值上的性能。
模型
Virtual ChIP-seq通过从公开的ChIP-seq实验、基因组保守性和所有基因表达与TF结合的相关性中学习来预测TF结合。主要通过学习转录组对TF结合影响的新表达,使用多层感知器整个表观基因组合基因组特征来实现。
该模型还可以在没有已知序列偏好的情况下准确预测一些DNA结合蛋白的位置。
染色质因子:受ChIP影响的因子。
模型预测了36种染色质因子在33种表观基因组学细胞类型上的结合。
数据:来自Cistrome DB和ENCODE的ChIPseq和来自癌细胞系百科全书(CCLE)和ENCODE的RNA-seq数据。除此之外还使用了DREAM挑战的针对31种染色质因子的chip-seq数据。
思想:
- 对于每个染色质因子,使用一个关联矩阵测量不同细胞类型中,不同的基因表达与先前收集的数据集中该染色质因子结合之间的相关性。
- 矩阵中的每个值对应在一个基因组结合处,染色质因子的ChIP-seq结合和一个基因的表达水平之间的皮尔逊相关性。
- 使用关联矩阵和rna-seq数据计算染色质因子的表达分数:关联矩阵中,该基因组结合的非NA值与基因的表达水平之间的Spearman相关性
方法
用于预测的数据
重叠的基因组bin:使用50bp滑动窗口的200bp基因组bin,排除和encode黑名单区域重叠的基因组,使用encode的GRCh38/hg38的数据。
染色质可及性:Cistrome DB ATAC-seq and DNase-seq的narrow peak文件。
基因组保守性:来自UCSC Genome Browser的GRCh38灵长类和胎盘哺乳动物7-way PhastCons基因组保护评分。
基因序列得分:使用FIMO从JASPAR 2016中搜索基序,以确定具有该TF序列基序的每个TF的结合位点。
RNA-seq:对每个基因下载带有rna-seq数据的encode表达矩阵,使用PharmacoGx检索了相似的CCLE RNA-seq数据,分析仅限于两个数据集共享的Ensembl基因id,并根据细胞类型对基因表达值进行排序。
表达评分:在N ≥ 5的训练细胞类型中,用匹配的ChIP-seq和RNA-seq数据为每个染色质因子建立了一个表达矩阵。
预测时,计算该细胞类型中每个基因组结合的表达分数,即为相同G=5000个基因的表达中,每一行都代表单个基因组结合的关联矩阵A的Spearman的 ρ \rho ρ值。接近1的表达分数表示高表达的基因在关联矩阵中具有高值,低表达的基因具有低值。接近1的表达分数表明高或低表达的基因在关联矩阵中具有相反的值。
建立表达矩阵:
- 将基因组分成M 100 bp的非重叠基因组仓;
- 创建了一个非负ChIP-seq矩阵 C ∈ R ≥ 0 M × N C ∈ R^{M×N}_{≥0} C∈R≥0M×N,使用MACS2为M个箱和N种细胞类型生成的重复窄峰文件中的信号平均值,并对该矩阵进行分位数标准化;
- 对C到C’进行行规范化,将每一行的值在0和1之间缩放;
- 确定了在N种细胞类型中具有最高方差的G = 5000个基因;
- 创建了一个表达矩阵 E ∈ R ∈ [ 0 , 1 ] N × G E ∈ R^{N×G} _{∈[0,1]} E∈R∈[0,1]N×G,其中包含N种细胞类型中G = 5000个基因中每个基因的行标准化表达等级;
- 对于每个结合物 i ∈ [ 1 , M ] i ∈ [1,M] i∈[1,M]和每个基因 g ∈ [ 1 , G ] g ∈ [1,G] g∈[1,G],计算该结合物 C i ′ C^′_i Ci′:的ChIP-seq数据和该基因E:,j在所有细胞类型中的表达等级之间的表观相关系数 A i , g A_{i,g} Ai,g,如果皮尔逊相关系数是不显著(p > 0.1),我们把 A i , g A_{i,g} Ai,g设为NA。这些系数构成一个关联矩阵 A ∈ ( R ∈ [ 1 , 1 ] ∪ N A ) M × G A ∈( R_{∈[1,1]}\cup{NA})^{M×G} A∈(R∈[1,1]∪NA)M×G。
训练、优化和基准
超参数的选择和训练:
- 输入矩阵:每一行对应200bp基因组窗口,列对应表达评分、染色质因子结合的先前证据、染色质可及性、基因组conservation、序列motif得分和HINT foot峰;
- 使用了50 bp位移的滑动基因组bins,在binging预测中提供了50bp的最大分辨率,提供了具有60620678行的代表GRCh38基因组装配中的每个bin的稀疏矩阵;
- 模型使用的系数矩阵具有4-11列,取决于可用基列序列的数量
多层感知器:全连接的前馈神经网络,每个基因组窗口的binding是和上下游窗口相独立的。使用自适应随机梯度下降,和200个样本训练。
超参数优化:4重交叉验证,包括激活函数,每层隐藏的单元数量、隐藏层的数量和L2正则化。
训练:一次对4条染色体中的3条进行迭代训练,并评估其余染色体的表现。经过4重交叉验证后,选择平均MCC最高的模型。
对于23个染色质因子,最优模型有10个隐层。对于另一组23个染色质因子,最佳模型有5个隐藏层。对于最后的17个染色质因子,最佳模型只有2个隐藏层。对于63个检测的染色质因子中的57个,表现最好的模型在每层都有100个隐藏单元。对于剩余的6个染色质因子,最优模型在每层有10-24个隐藏单元。
对于不同的染色质因子,最佳的激活函数是不同的。
隐藏层数、隐藏单元数或激活函数与模型性能之间没有显著相关性。
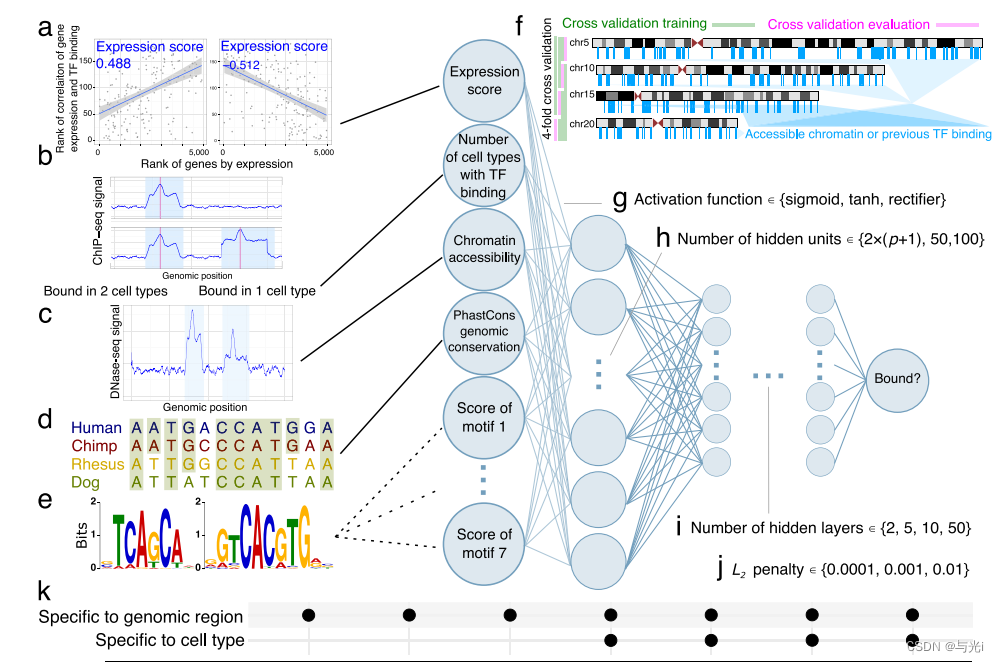
图1 模型结构
基准测试:使用R precrec软件包来计算auPR和auROC,精确召回(PR)曲线比接收器操作特性(ROC)曲线更好地评估了二进制分类器在不平衡测试数据上的性能。也在GRCh37DERAM挑战数据上训练和验证了虚模型。
结论
Virtual ChIP-seq使用了全连接神经网络,整合了转录组transcriptome、染色质可及性、基因组背景数据,能够预测TF的结合,并且也能够正确预测训练细胞类型中不存在的新peak。
与DREAM Challenge数据集相比,本文的数据集使用Cistrome DB和ENCODE,允许训练和验证预测更广泛的63种染色质因子结合的模型,具体可以预测33种不同路线图组织类型中36种高可信度染色质因子的结合。