转录组之质量控制(FastQC)
本文简介:这篇文章是经过了自己学习实践出来的,参考了很多资料,如若有大佬能指出错误,我将感激不敬,有错误,请在评论区留言,谢谢。
更新日期:2023年7月4号
本文知乎地址: 转录组之质量控制(FastQC)[学习笔记通俗易懂版]
fastqc软件简介
FastQC是一个用于高通量序列数据的质量控制程序。FastQC可以读取并分析多种格式的序列数据,并且可以以交互的形式来检查几种不同的质量结果,或者创建一个可以集成到自动分析流程中的报告。该软件生成的结果是html格式文件,使用可以使用浏览器打开,非常便捷。
fastqc 软件的安装(Linux环境,不提供windows)
fastqc软件需要依靠java环境,我这里提供两种方式安装。
一、使用安装包安装,在fastqc官网下载安装包,但是这种方法需要预先安装Java环境(Jdk),如果你不确定是否安装了Java环境,请打开你的服务器终端输入java -version,如果有java版本显示,则说明安装过了Java,如没有安装,则会返回Java未找到。(安装过Java的机器如下图)
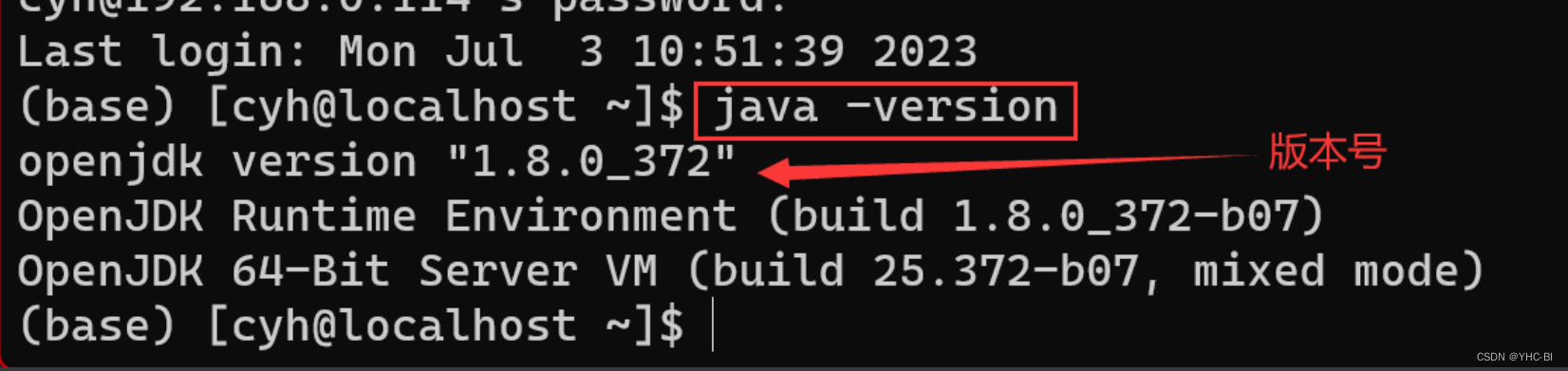
如果没有安装Java环境,则需要预先安装java环境,安装步骤如下:
The fastQC software need java enviroment,so we must system had install java ,if not ,that we start to download java and dispose java enviroment
使用yum源安装:
yum install java #使用yum源安装
yum安装Java后,配置Java环境:
- 编辑
/etc/profile文件
vim /etc/profile #edit this file and input the following content
- 文件末尾输入以下内容,可以直接抄。
# java enviroment
JAVA_HOME=/usr/lib/jvm/jre-1.8.0-openjdk-1.8.0.372.b07-1.el7_9.x86_64
PATH=$PATH:$JAVA_HOME/bin
CLASSPATH=.:$JAVA_HOME/lib/dt.jar:$JAVA_HOME/lib/tools.jar
export JAVA_HOME CLASSPATH PATH
-
重启
/etc/profile文件source /etc/profile #重启文件 -
检查,使用查看Java版本的命令检查环境是否配置成功——
java -version若不成功,请重新检查。如果看了抄了我的还是不成功,请看/usr/lib/jvm文件是否使用yum成功安装了java。
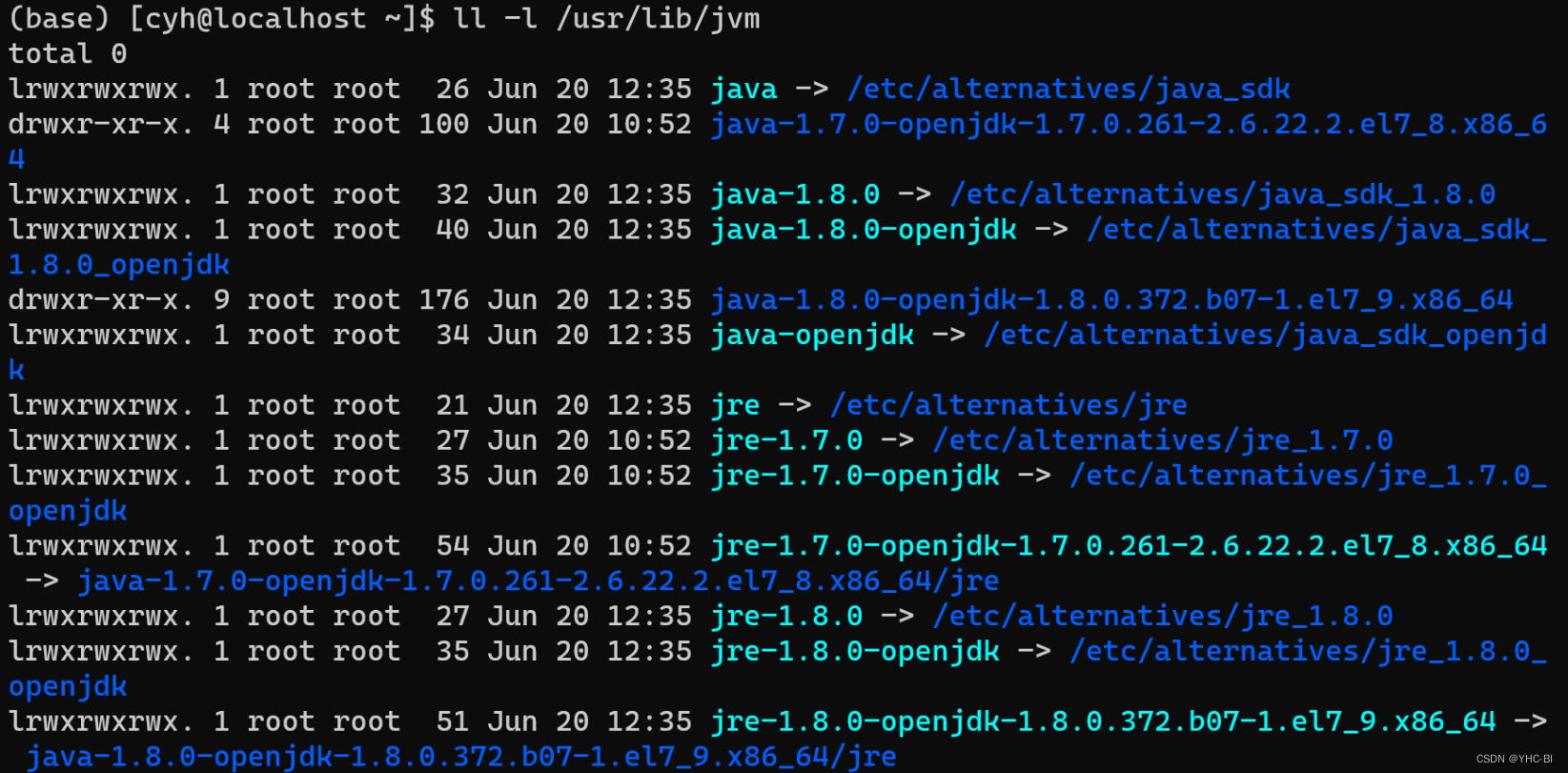
Java软件使用yum源是直接安装在了/usr/lib/jvm中。我们可以查看以下这个文件:
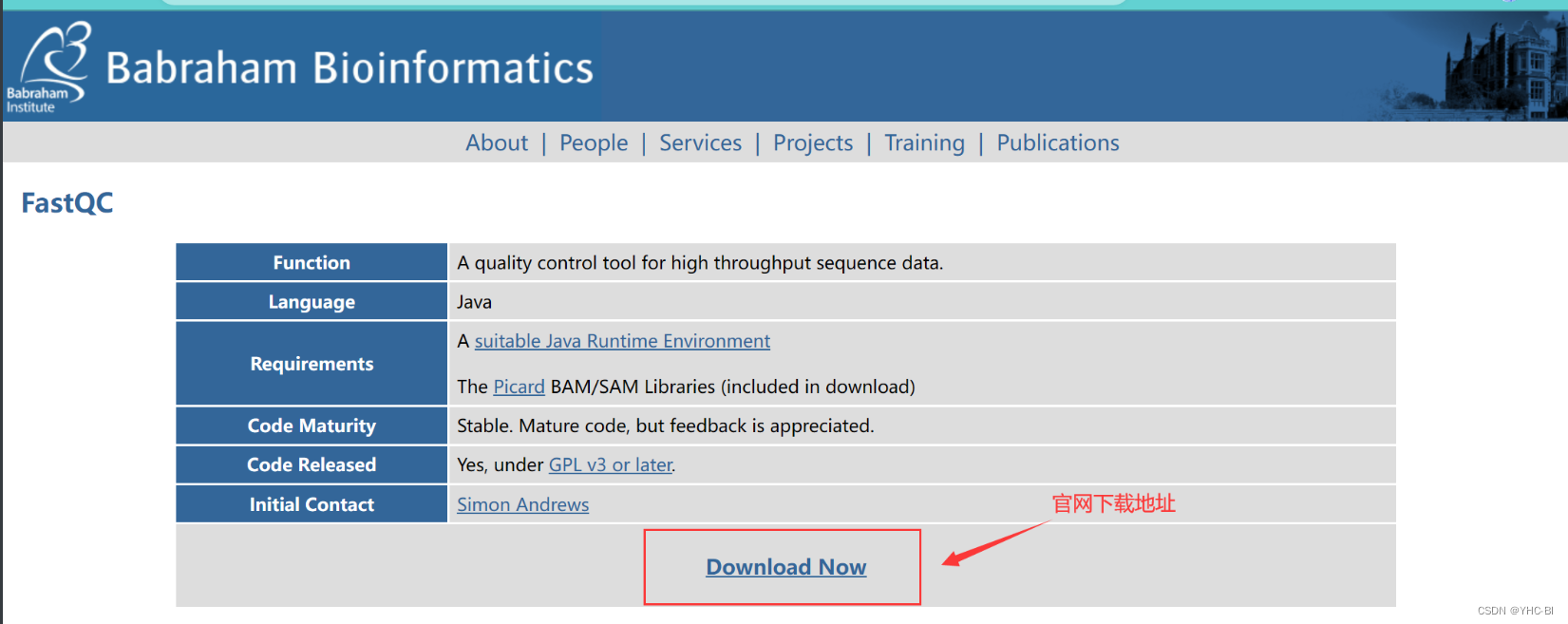
安装成功Jdk后我们浏览器搜索fastqc,找到其官网,我一般从GitHub下包。下面我举个例子:(请下载最新版,我的版本可能在你看的时候过时了)
官网地址(Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data)
官网页面,可以参考下图标记位置下载fastqc
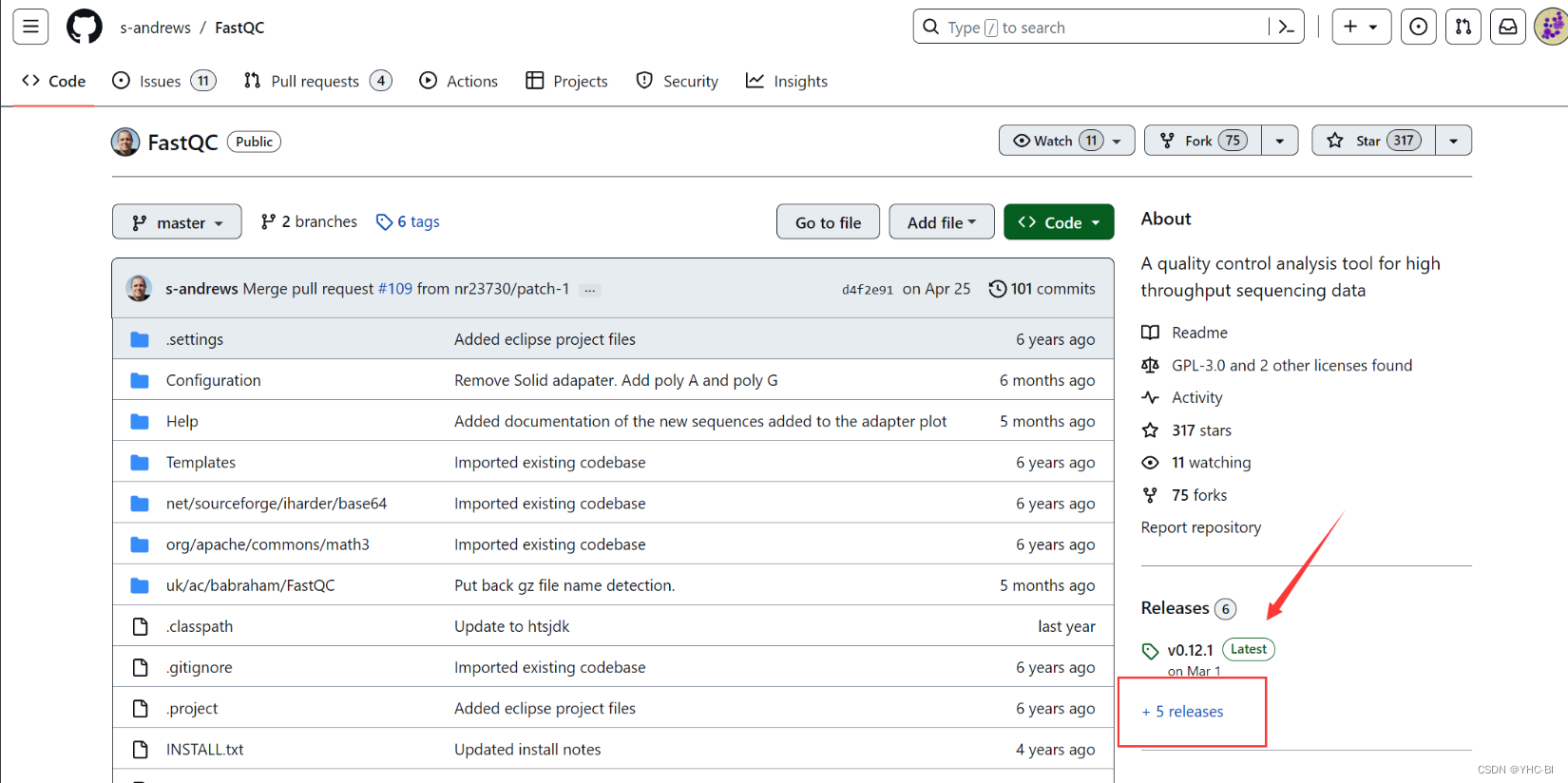
我一般从GitHub下载,地址(Releases · s-andrews/FastQC (github.com))
wget命令下载包(官网)
#download fastqc paskage
wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.12.1.zip
- 配置fastqc环境,在
~/.bashrc文件(或者在其他文件配置)末尾加入以下代码:
#dispose enviroment
# fastQC
#格式如下:
#export PATH="包的路径:$PATH"
export PATH="/home/cyh/biosoft/FastQC: $PATH" #路径不一样,请勿完全抄,仅供参考
- 重启文件:
# reboot file
source ~/.bashrc
- 检查是否可行,help一下,如果有帮助信息,则成功,否者重新检查
fastqc --help
二、上述我们使用了安装包安装,还有一种方法可以不用特意安装Java,我们可以使用生物学常用的工具miniconda3,推荐使用,应为很多生物学软件它都有,很方便。
- 安装
miniconda3,从官网下包安装,自己上官网找到下载地址,我这里使用wget命令下载安装包,同样推荐下最新版。下载后可以看到是个.sh脚本。
wget https://repo.anaconda.com/miniconda/Miniconda3-py310_23.1.0-1-Linux-x86_64.sh #下载
- 运行
.sh脚本。
bash Miniconda3-py310_23.1.0-1-Linux-x86_64.sh #通过bash安装.sh安装包
- 它会自动配置环境,不要动它。直接重启
/.bashrc文件。
source /.bashrc #重启.bashr
- 重启文件后,如果命令行开头有base显示,这表示成功,如:
(base) [cyh@localhost ~]$
- 安装
miniconda3后,通过该软件安装fastqc,下面有两条命令,第二条命令最简单,第一条命令需要选择你期望的版本。(建议最新版)
conda search bioconda fastqc #查询conda中是否有fastqc安装包,并返回所有版本
conda install -c bioconda fastqc #默认安装最新版fastqc
- 检查安装是否成功,出现软件帮助信息表示成功
fastqc --help
到此两种方式安装fastqc软件完成,若同学你未安装成功,且检查后无误,请找其他安装教程。
FastQC软件的使用
FastQC 支持 fastq、gzip 压缩的 fastq、SAM、BAM 等格式,在不指定文件类型的情况下,FastQC 会根据文件的名字来推测文件的类型: 以 .sam 或者 .bam 结尾的文件会被当作 SAM/BAM 文件来打开,并统计 mapped 和 unmapped reads 在内的所有 reads;其它的文件类型则被当作 fastq 格式打开。 其使用语法为:
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] [-t threads] seqfile1 .. seqfileN
[-o output dir]:指定FastQC输出结果的目录。默认情况下,结果将保存在当前工作目录中。[--(no)extract]:设置是否解压缩输入文件。FastQC可以对gzipped或bzipped的文件进行处理,解压缩文件能够提供更准确的结果。该选项默认为自动解压缩。[-f fastq|bam|sam]:指定输入文件的格式。可以选择fastq(默认),bam或sam。根据输入文件的类型选择合适的格式。[-c contaminant file]:指定一个污染物文件,用于检测和过滤掉可能存在的污染物序列。该文件包含了可能出现在样本中的污染物序列。[-t threads]:指定使用的线程数。FastQC可以利用多个线程加快分析速度。默认为单线程处理。seqfile1 .. seqfileN:输入的序列文件列表。可以指定一个或多个文件进行分析。
命令很简单,举例如下:
fastqc -t 4 -o /home/cyh/*.fq /home/cyh/result/
例子数据存放在 /home/cyh/中,输出到指定文件夹 /home/cyh/result/,使用了4个线程。
一个数据会生成两个结果,一个是HTML文件,一个.zip文件,我们需要查看的是HTML文件。
(base) [cyh@localhost ~]$ ll -l /home/cyh/result
-rw-rw-r--. 1 cyh cyh 239289 Mar 31 15:53 xxx_fastqc.html # html 文件
-rw-rw-r--. 1 cyh cyh 239289 Mar 31 15:53 xxx_fastqc.zip
对于生成的结果,我们只需查看html文件。
FastQC报告解读目的
大多数测序仪在其分析流程中会生成一个质量控制报告,但这通常只关注由测序仪本身产生的问题。FastQC旨在提供一个质量控制报告,该报告可以发现源于测序仪器或起始文库材料的问题。
基本生信分析流程(例如RNA-Seq)的第一步是对测序的原始数据fastq文件进行质量评估,做简单的质量控制检查,以确保原始数据的质量良好,且您的数据中没有可能影响到有用应用的问题或偏差。以便后续组装过程中丢弃低质量的片段,减少因测序错误而产生的影响。
THML结果解读
可参考地址:FastQC_Manual.pdf (missouri.edu)
我们使用浏览器打开html文件,对于生成的结果,我们只需查看html文件。
对于FastQC Report ,主要分为11个部分(有些生成出的结果不一,与你的质量有关),如图:
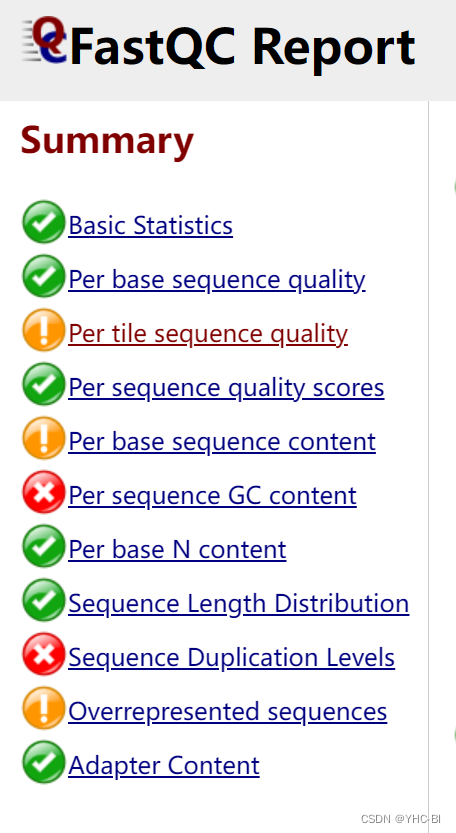
接下来我们来一一解读FastQC Report
-
Basic Statistics 部分:
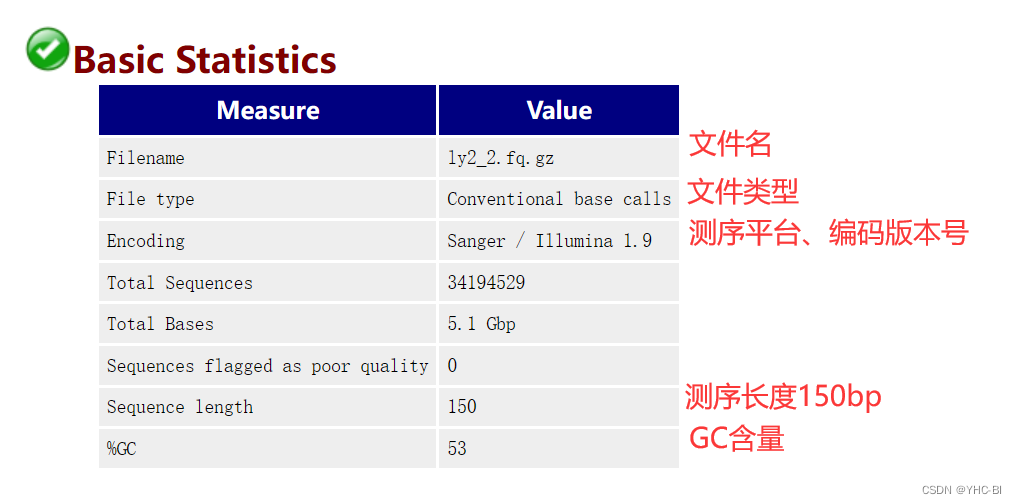
该部分包括Filename(文件名)、File type(文件类型)、Encoding(测序平台)、Total Sequences(总序列数)、Sequences flagged as poor quality(低质量测序碱基数)、Sequence length(序列长度,给出最短和最长的序列长度,若是所有序列长度一致,则只给出一个值)和%GC(所有序列总的GC含量)。%GC:是我们需要重点关注的一个指标,这个值表示的是整体序列中的GC含量,这个数值一般是物种特异的,比如人类细胞就是42%左右。如果测序原始数据的GC含量远远偏离这个比例,说明测序数据存在一定偏好性,如果直接用测序数据,会影响后续的CNV和变异检测的分析。 -
Per base sequence quality部分
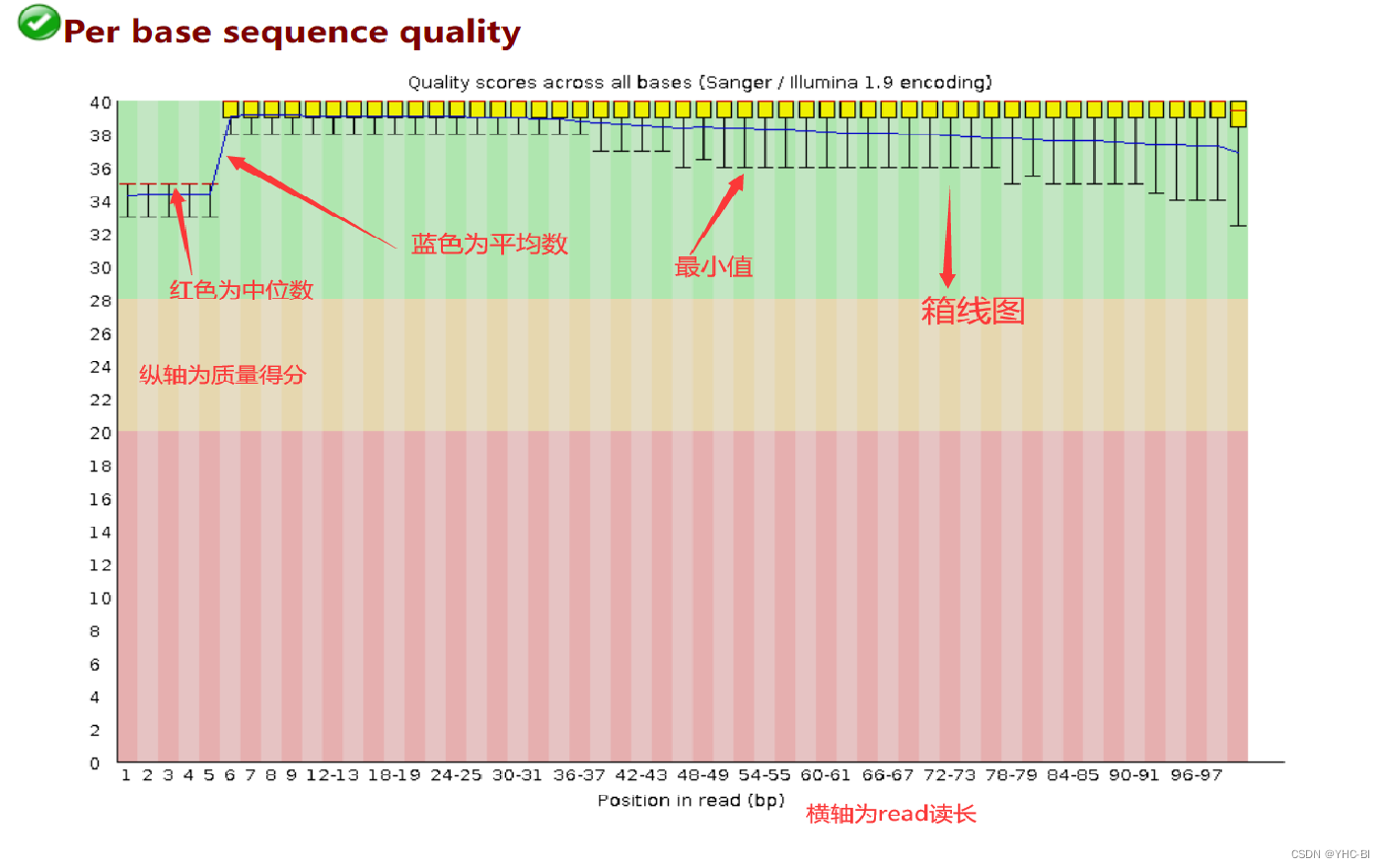
该部需要注意,图片上标注了各部分含义,箱线图应该位于蓝色部分就是合格的,黄色部分出警告,红色部分不合格;
这是 read length = 100 的数据,横轴为read位置,纵轴是quality。quality = -10*log10( p),p为测错的概率。根据quality给出质量结果:正常区间(28 - 40),警告区间(20-28),错误区间(0-20)。
比如,当read的某一位置的p=0.01,quality=20,那么它就处于错误区间。
本部分标准如下:如果任何一个位置的下四分位数小于10或者中位数小于25,会显示“警告”;如果任何一个位置的下四分位数小于5或者中位数小于20,会显示“不合格 -
Per tile sequence quality部分
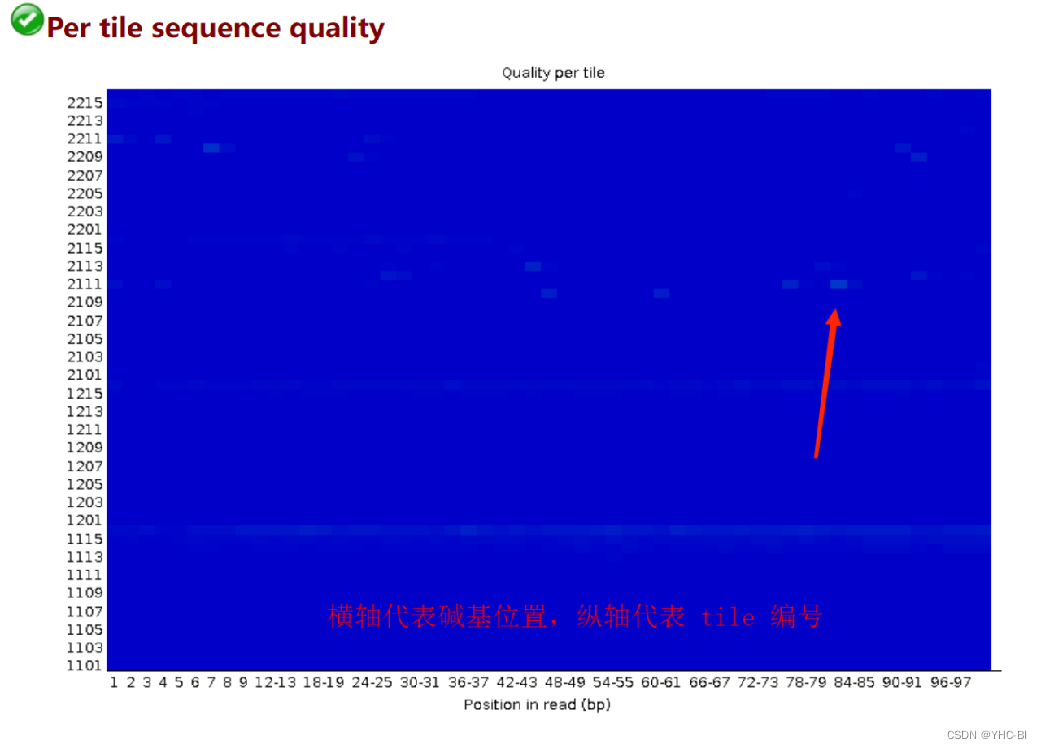
该图片质量很好,下面展示局部不好的(因为本人没有不好的图)

tile:每一次测序荧光扫描的最小单位。
图中的颜色是从冷色调到暖色调的渐变,冷色调表示这个 tile 在这个位置上的质量值高于所有 tile 在这个位置上的平均质量值,暖色调表示这个 tile 的在这个位置上的质量值比其它 tiles 要差;一个非常好的结果,整张图都应该是蓝色,横轴代表101个碱基的位置;纵轴是tail的Index编号。
检查reads中每一个碱基位置在不同的测序小孔之间的偏离度,蓝色表示低于平均偏离度,偏离度小,质量好;越红表示偏离平均质量越多,质量也越差。如果出现质量问题可能是短暂的,如有气泡产生,也可能是长期的,如在某一小孔中存在残骸,问题不大。
合格标准:
如果任何 tile 的平均质量值与这个位置上所有tiles的平均质量值相差2以上会显示“警告”,如果任何 tile 的平均质量值与这个位置上所有 tiles 的平均质量值相差5以上会显示“不合格” -
Per sequence quality scores部分
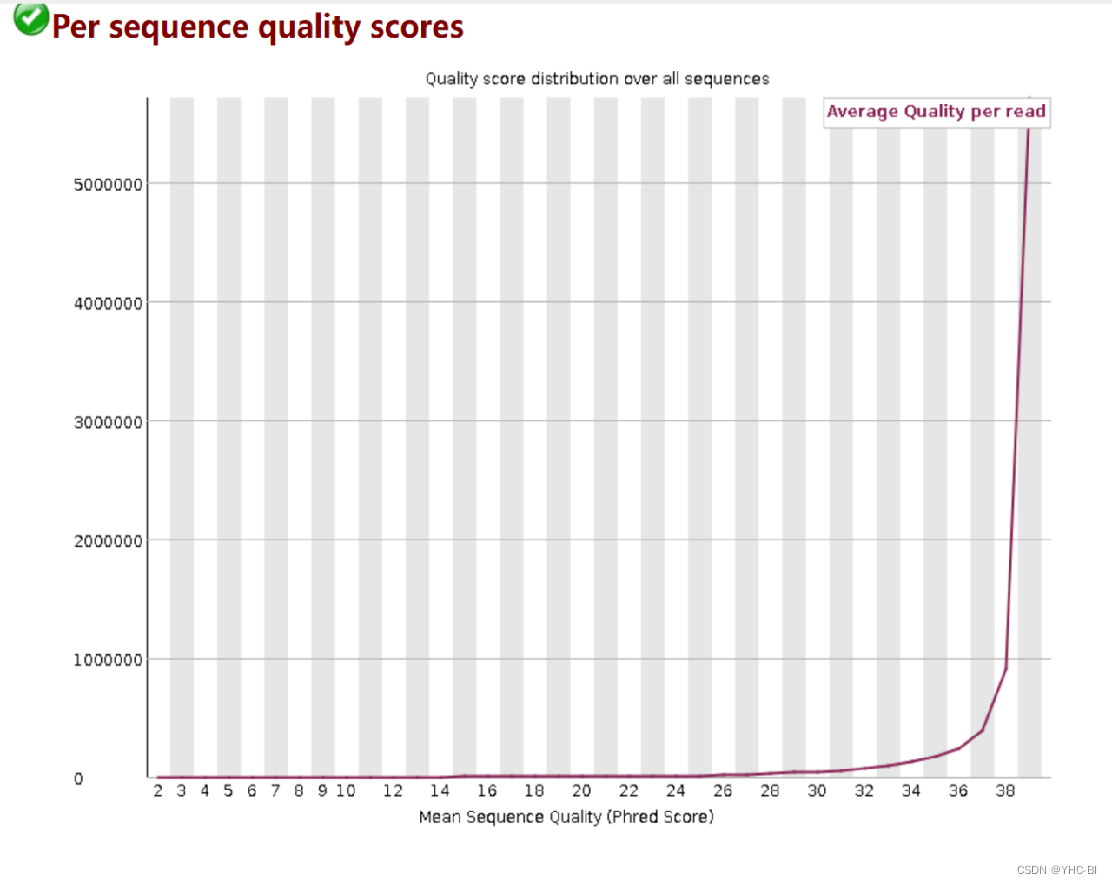
序列平均质量频数。x轴是平均碱基质量值,y轴是平均碱基质量值对应的reads数。
如果最高峰的质量值小于27( 错误率0.2%)则会显示“警告”,如果最高峰的质量值小于20(错误率 1 %)则会显示“不合格”。上图显示本数据很好。
低质量的会有两或多个峰 -
Per base sequence content部分
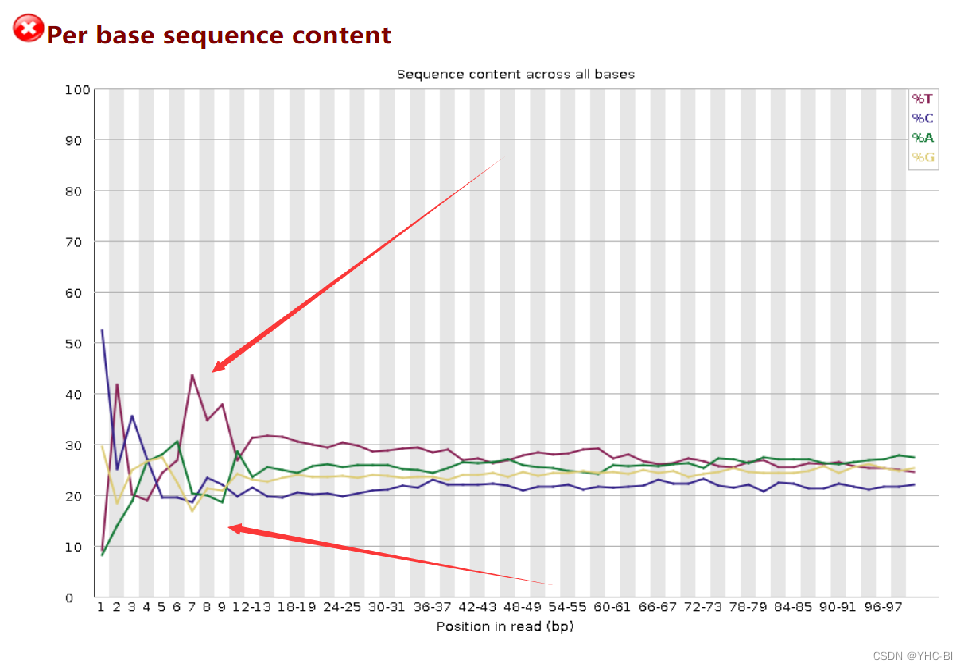
图中横轴为碱基位置,纵轴为碱基组成比例;
一个完全随机的文库内每个位置上 4 种碱基的比例应该大致相同,因此图中的四条线应该相互平行且接近;
在 reads 开头出现碱基组成偏离往往是我们的建库操作造成的,比如建 GBS 文库时在 reads 开头加了 barcode;barcode 的碱基组成不是均一的,酶切位点的碱基组成是固定不变的,这样会造成明显的碱基组成偏离;或者是测序仪状态不稳定。
在 reads 结尾出现的碱基组成偏离,往往是测序接头的污染造成的,所以在开头会出现不规则波动。目前该问题好像没解决,但是可以减弱,比如减少接头污染。
在序列前12~15个碱基,可以使用剪切软件切除,切除多少个需要看你的数据,比如上图的结果显示就可以切12个或13个。
合格标准:
如果任何一个位置上的 A 和 T 之间或者 G 和 C 之间的比例相差 10 % 以上则报“警告”,任何一个位置上的 A 和 T 之间或者 G 和 C 之间的比例相差 20 % 以上则报“不合格” -
Per sequence GC content部分
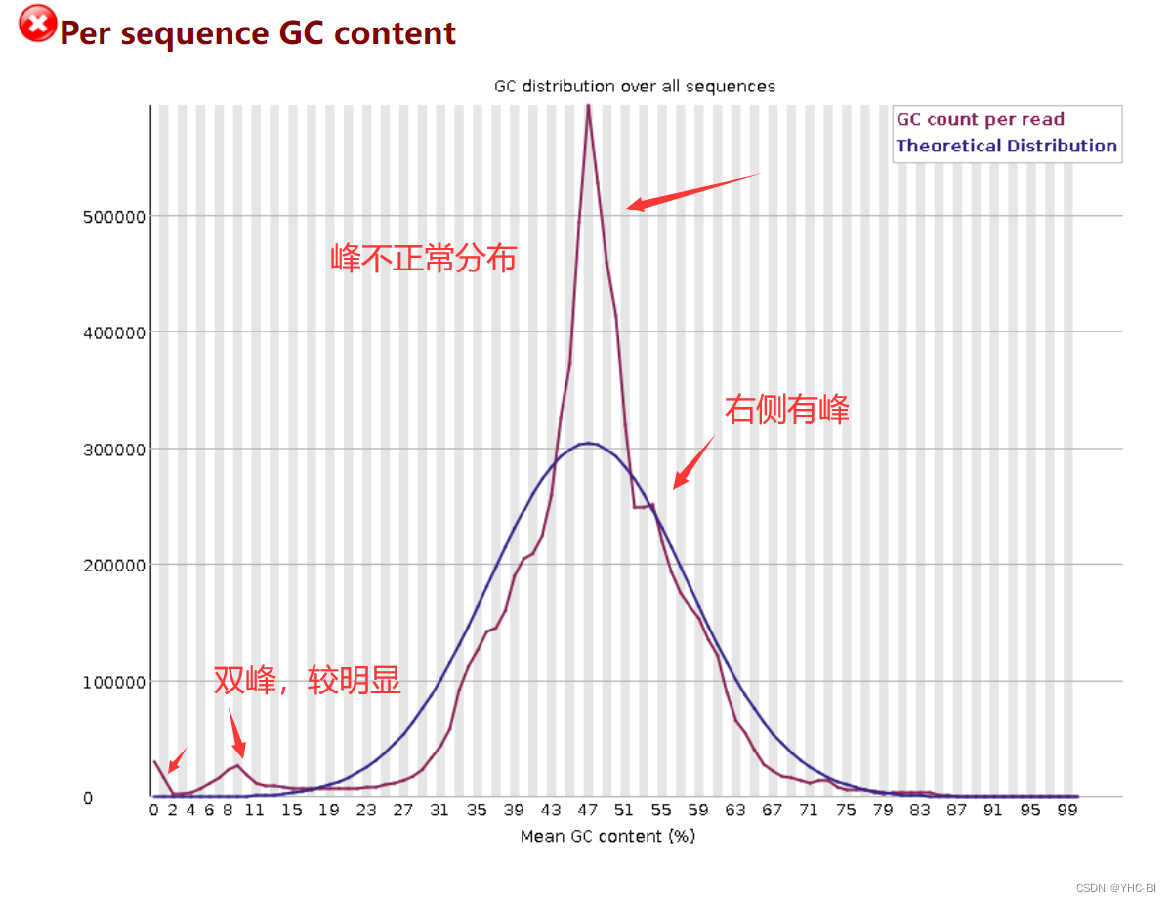
上图的标记的双峰是我认为有影响的,请不要参考。
这部分表示统计每个序列GC含量的频数。红色线是实际值,蓝色线是理论值。峰值位点对应着总体GC含量。在一个正常的随机文库中,GC 含量的分布应接近正态分布,且中心的峰值和所测基因组的 GC 含量一致。由于软件并不知道所测物种真实的 GC 含量,图中的理论分布是基于所测数据计算得来的;红色线于蓝色线越接近越好,越接近测序越好,越可靠。
如果出现不正常的尖峰分布 (如本图),则说明文库可能有污染(如果是接头的污染,那么在Overrepresented Sequences那部分结果还会得到提示),或者存在其它形式的偏选;
合格标准:
如果偏离理论分布的 reads 数超过总 reads 数的 15 % 则报“警告”,如果偏离理论分布的 reads 数超过总 reads 数的 30 % 则报“不合格”。 -
Per base N content部分
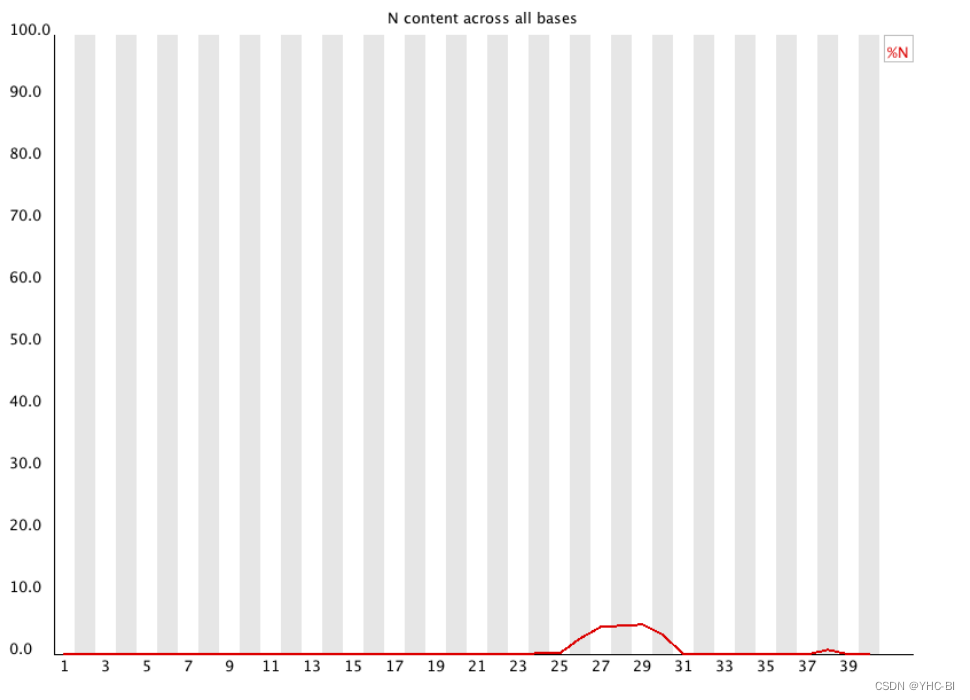
(上述图片来源于https://dnacore.missouri.edu/PDF/FastQC_Manual.pdf,其中25至31部分含有N值)
在测序仪工作过程中,如果不能正常完成某个碱基的 calling,将会以 N 来表示这个位置的碱基,而不是 A、T、C、G;
有时在序列中会出现较低比例的 Ns,尤其是靠近序列末端的位置,这说明系统不能正常的 call 这部分碱基;出现一定比例的 Ns 最常见的原因是普遍出现的质量丢失 (a general loss of quality),这种情况可结合其它部分的结果来综合判断;另一种常见的现象是文库整体上的测序质量较高,但 reads 开头出现较高比例的 N,这可能是由于文库的碱基组成偏离的比较严重,测序仪不能给出正确的 call,这种情况可以结合 per-base sequence content 的结果来判断;该N的含量越小越好,为0是最最最理想的。
合格标准:
如果任何一个位置 N 的比例大于 5 % 则报“警告”,大于 20 % 则报“失败”。 -
Sequence Length Distribution部分
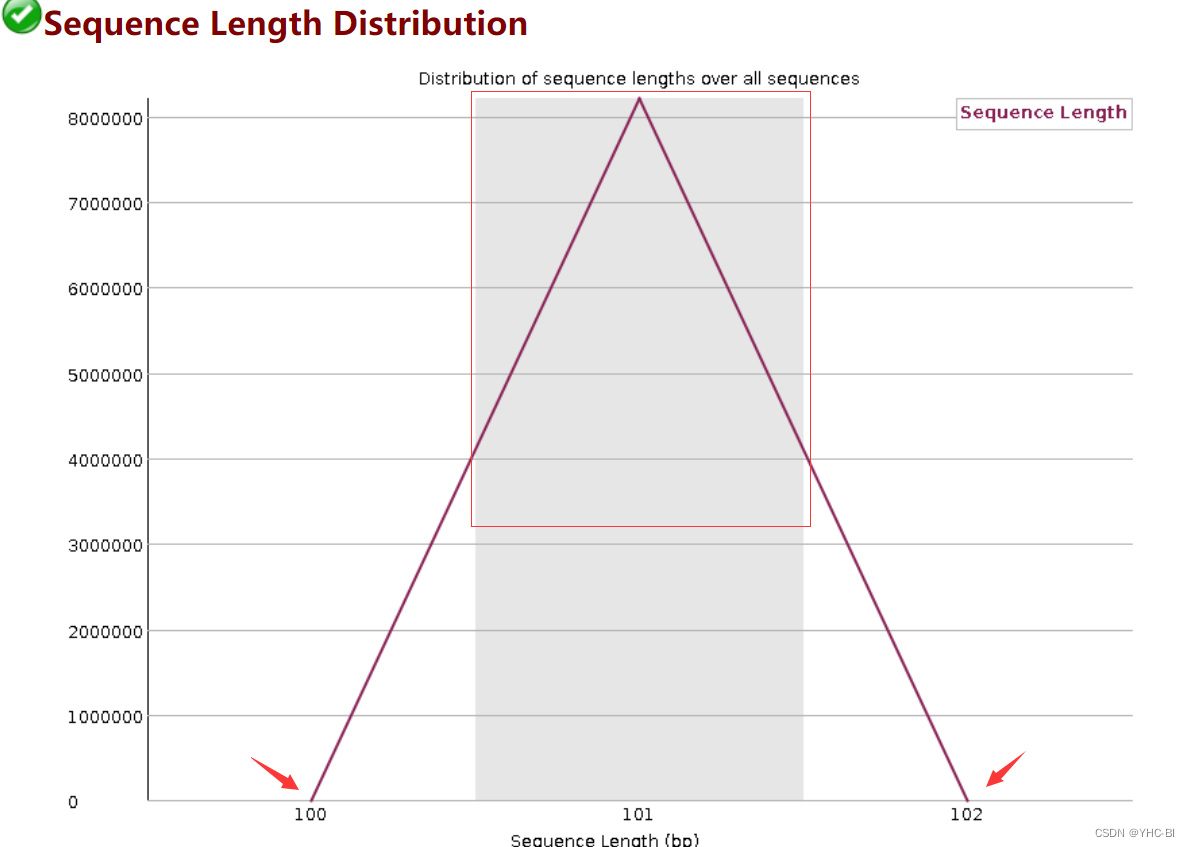
该部分是序列长度分布情况,上图例子显示,整个数据序列的长度分布于100~102之间。测序仪出来的原始 reads 通常是均一长度的,但经过质控软件等处理过的数据则不然;每个序列长度理论上完全一样的。
合格标准:
当 reads 长度不一致时报“警告”,当有长度为 0 的 reads 时则报“不合格”。 -
Sequence Duplication Level部分
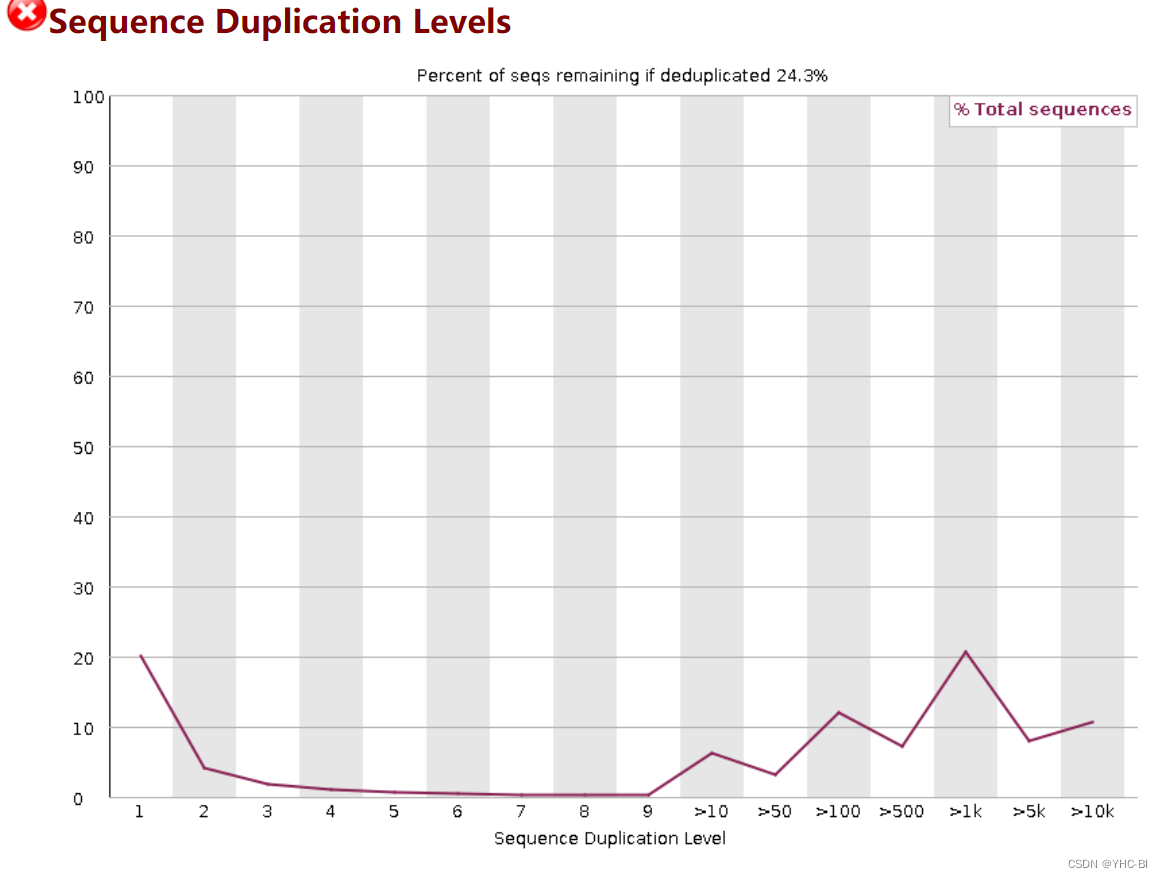
横坐标为重复(duplication)的次数,纵坐标为reads的数目,以unique reads的总数作为100%
合格标准:
如果非唯一序列占总数的20%以上,该模块将发出警告。
如果非独特的序列占总数的50%以上,该模块将发出一个错误。
以下解释来源于
https://dnacore.missouri.edu/PDF/FastQC_Manual.pdf
一个多样化的文库中,大多数序列在最终集合中只出现一次。一个低水平的 重复率低可能表明目标序列的覆盖率很高,但是高重复率更可能表明某种富集偏差(如PCR过量)。重复程度高更有可能表明某种富集偏差(如PCR过度扩增)。该模块计算了序列集中每个序列的重复程度,并创建了一个 图显示具有不同重复程度的序列的相对数量。
为了减少该模块的内存需求,只分析每个文件中前20万个序列中出现的序列。分析每个文件中的前200,000个序列,但这应该足以对整个文件的重复程度有一个好的印象。但这应该足以对整个文件中的重复程度有一个好的印象。每个序列都被追踪到 每个序列都被追踪到文件的末尾,以提供总体重复水平的代表性计数。为了减少 为了减少最终图表中的信息量,任何有10个以上重复的序列都被放入10个重复的序列中。重复的序列都被放入10个重复的类别中–因此,在这个最终类别中看到一个小的 所以在这个最终类别中出现小幅上升是很正常的。如果你在这个最后类别中看到一个大的上升,那么它意味着你有 有大量的重复程度非常高的序列。
-
Overrepresented sequences部分
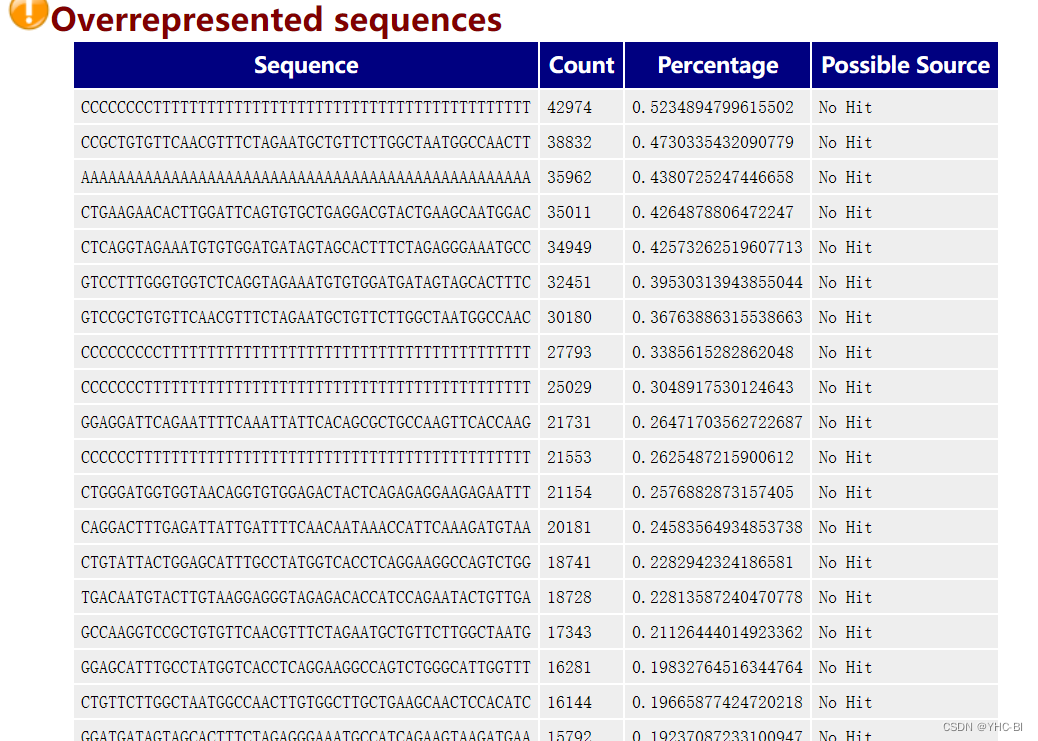
它展示了长度至少20bp,数量占总数0.1%以上的reads碱基组成,它可以帮助判断污染(比如:载体、接头序列)。(某一条序列占总序列的 0.1%,则被鉴定为过表达序列。)
如果上面的GC含量分布图 "挂了 ",这个表可以帮助我们判断来源,如果是已知的载体或者接头,它会列出来;如果不是,可以复制序列去blast(这好像是个软件我没用过( ~ _ ~ )。
标准如下:
当发现超过总reads数0.1%的reads时报“WARN”,当发现超过总reads数1%的reads时报“FALL”。 -
Adapter Content部分
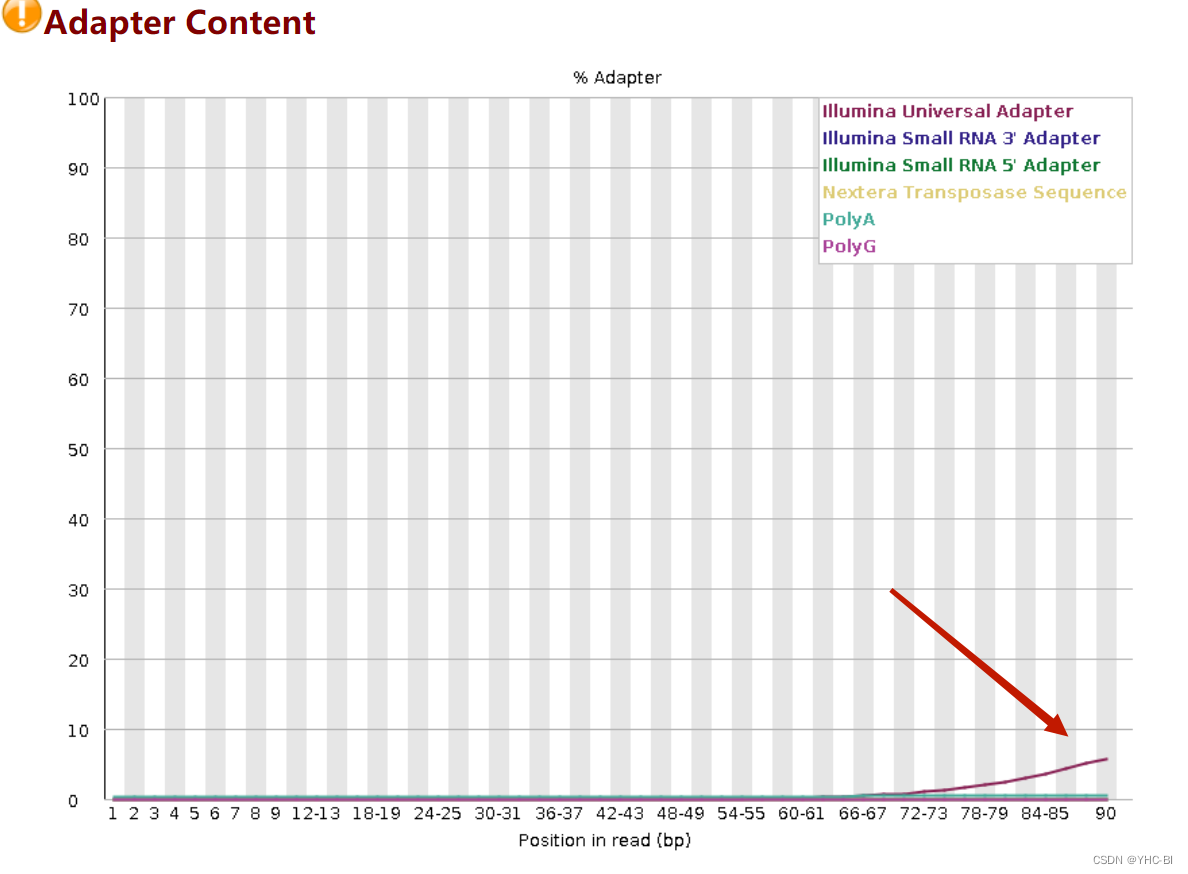
这部分衡量的是序列中两端adapter的情况
如果在当时fastqc分析的时候-a选项没有内容,则默认使用图例中的四种通用adapter序列进行统计。本例中adapter没有去除,在72~90部分含有曲线翘起,在后续分析的时候需要先使用去接头软件进行去接头。
FastQC报告解读结束。
到此,本文内容结束,这篇文章是经过了自己学习实践出来的,参考了很多资料,如若有大佬能指出错误,我将感激不敬,有错误,请在评论区留言,谢谢。