Benchmarking atlas-level data integration in single-cell genomics
January 2022 发在《Nature Methods》
多批次单细胞(带标签)整合分析流程主要包括整合–去批次效应–保留生物学变异信息
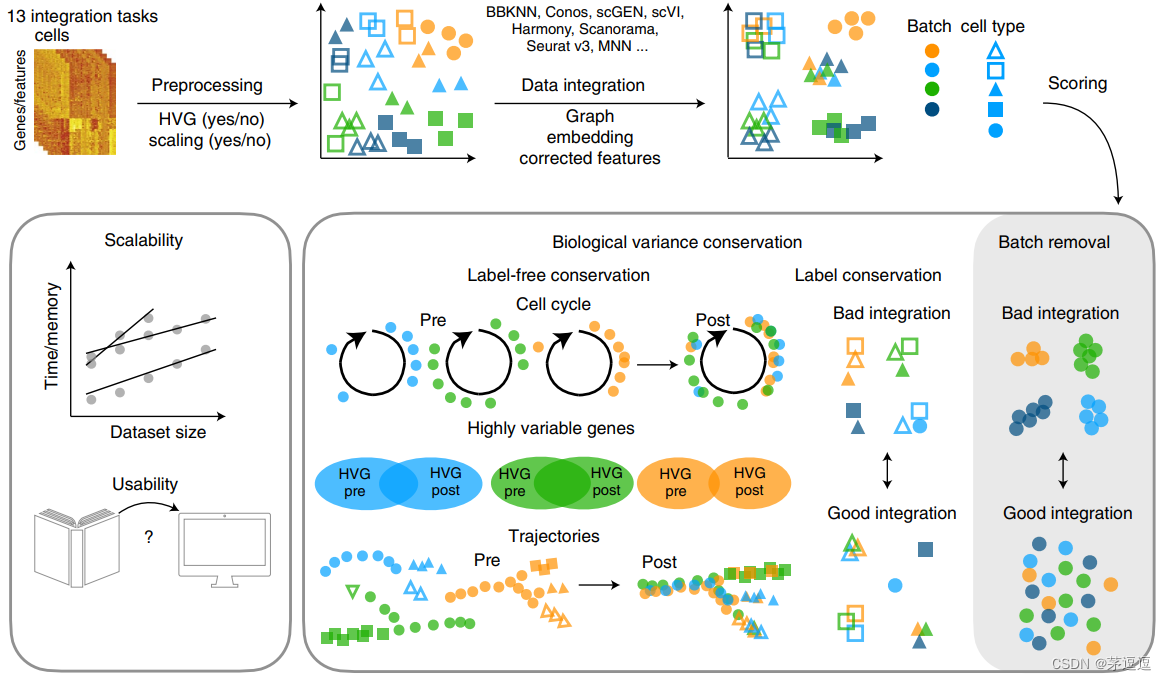
论文摘要
Single-cell atlases often include samples that span locations, laboratories and conditions, leading to complex, nested batch
effects in data. Thus, joint analysis of atlas datasets requires reliable data integration. To guide integration method choice, we
benchmarked 68 method and preprocessing combinations on 85 batches of gene expression, chromatin accessibility and simulation data from 23 publications, altogether representing >1.2 million cells distributed in 13 atlas-level integration tasks. We
evaluated methods according to scalability, usability and their ability to remove batch effects while retaining biological variation
using 14 evaluation metrics. We show that highly variable gene selection improves the performance of data integration methods, whereas scaling pushes methods to prioritize batch removal over conservation of biological variation. Overall, scANVI,
Scanorama, scVI and scGen perform well, particularly on complex integration tasks, while single-cell ATAC-sequencing integration performance is strongly affected by choice of feature space. Our freely available Python module and benchmarking pipeline
can identify optimal data integration methods for new data, benchmark new methods and improve method development.
我简单翻译一下:
单细胞测序数据往往来自于不同的地点、不同的实验室和条件的样本,从而导致复杂的数据批次效应。因此,数据集的联合分析需要可靠的数据集成。为了指导集成方法的选择,我们对来自 23 篇出版物的 85 批基因表达谱数据、染色质可及性数据和模拟数据,分别使用68 种不同的方法和预处理组合进行了测评,我们的工作总共涉及13 个整合数据任务中、 >120 万个细胞。我们根据可扩展性、可用性及其在保留生物变异的同时消除批次效应的能力评估方法的好坏优良,且使用了 14 个评估指标去评价他。
我们的研究表明,高变的基因选择提高数据集成方法的性能,而数据标准化往往只是优先考虑批次效应的去除而不是生物变异性的保护。总的来说,scANVI,Scanorama、scVI 和 scGen 表现良好,特别是在复杂的集成任务上,而单细胞 ATAC 测序集成性能受特征空间选择的强烈影响。我们免费提供的 Python 模块和测试管道可以确定新数据的最佳数据集成方法,对标新方法并改进方法开发。
测评结果
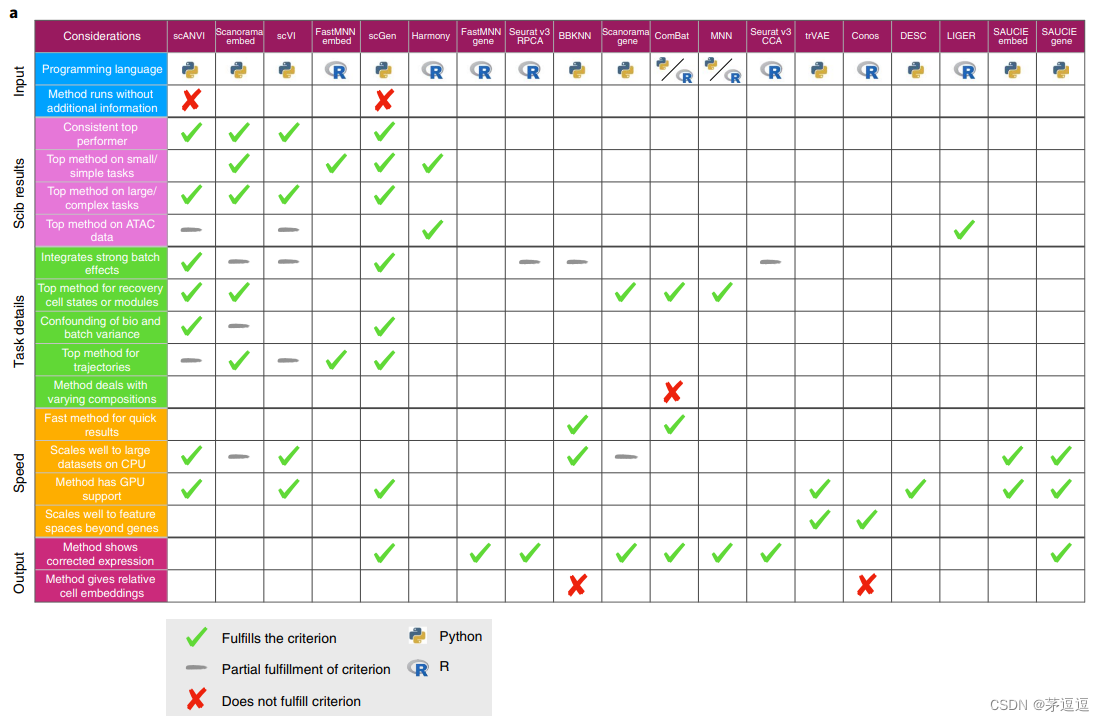
总结
这篇文章很有参考价值,在做批次去除效应的适合应该参考