一、数据准备
10X单细胞转录组理论上有3个文件才能被读入R进行seurat分析,分别是barcodes.tsv 、 genes.tsv和matrix.mtx,文件barcodes.tsv 和 genes.tsv,就是表达矩阵的行名和列名
pbmc.data <- Read10X(data.dir = "hg19")
#Load the PBMC dataset,创建Seurat对象,counts为读取的源文件,project为Seurat对象想保存的文件名,
#可以加上限定条件:min.cells为组织中分离的最少细胞数,min.features为一个细胞中测出的最少的基因数量
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
#查看pbmc中细胞数量的3个方法
pbmc
## An object of class Seurat
## 13714 features across 2700 samples within 1 assay ,细胞数量是2700个
## Active assay: RNA (13714 features, 0 variable features)
ncol(pbmc)
## [1] 2700
ncol(pbmc.data)文件解读
genes.tsv文件(有时也叫features.tsv文件)
文件内容:有两列,第一列为基因ID,第二列为基因Symbol ID,区分各个基因。

barcodes.tsv文件
文件内容:有一列,内容为测序时为了区分各个细胞的标记信息,称为Barcodes

matrix.mtx
文件内容:有三列,数字的第一行是测序的汇总信息。第一行的第一个为测序的总基因数(说明genes.tsv文件中有32738行),第二个为测序的总细胞数(说明barcodes.tsv文件中有2700行),第三个为测序的总reads数。 除第一行,其余数字记录矩阵信息,前两列代表坐标,第三列表示具体某个细胞某个基因的表达量。

表达量矩阵
根据上述三个文件可以得到表达量的稀疏矩阵。
下游处理的时候,一定要保证这3个文件同时存在,而且在同一个文件夹下面,每一个样本都是3个文件,每一个样本都是同样的代码处理。
二、一般流程
(一)数据前处理:质控和数据过滤
1.基于QC度量的细胞选择与筛选(即质控)
2.数据标化与缩放(即数据标准化)
3.高度可变特征的检测(特征性基因的选择)
进行质控
我们提到单细胞数据质控的时候,⼀般是指细胞的过滤,其实是从⼀个barcode X gene矩阵中过滤掉⼀部分不是细胞的barcode,如细胞碎⽚,双细胞,死细胞等。这三类barcode的特征可以通过其对应的基因表达情况来描述:nCount(总基因表达数)、nFeature(总基因数)、percent.HB(红细胞基因表达⽐例)、percent.MT(线粒体基因表达⽐例)。nCount和nFeature过⾼可能是双细胞,过低可能是细胞碎⽚。percent.HB刻画红细胞⽐例,percent.MT刻画细胞状态,值过⾼可能是濒临死亡的细胞
PercentageFeatureSet 函数是根据counts总数相除算的打分:该基因集的counts总和/所有基因的counts总和。
基因集占的百分比 = 分子 / 分母 * 100;
分子: 指定基因的 counts种总和,在这次分析中是指细胞的线粒体基因转录本数
分母: 从 meta.data 获取 nCount_RNA 列,就是每个cell中所有基因的 counts总和
这个函数的意思是,每个细胞的线粒体的基因数/细胞总基因数的百分比,将作为一列数据加到pbmc的metadata中,这一列的列名为“percent.mt"
#进行质控,计算每个细胞的线粒体基因转录本数的百分比(%),使用[[ ]] 操作符存放到metadata中;
#分析的时候要确认好物种,人用^MT-如果是小鼠的,要用mt
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
#在创建对象 CreateSeuratObject() 的过程中,会自动计算细胞中独特基因与总基因数目,可以在目标对象中找到
head([email protected],5)
orig.ident nCount_RNA nFeature_RNA percent.mt
AAACATACAACCAC-1 pbmc3k 2419 779 3.0177759
AAACATTGAGCTAC-1 pbmc3k 4903 1352 3.7935958
AAACATTGATCAGC-1 pbmc3k 3147 1129 0.8897363
AAACCGTGCTTCCG-1 pbmc3k 2639 960 1.7430845
AAACCGTGTATGCG-1 pbmc3k 980 521 1.2244898进行质控前

进行质控后(小提琴图)
#(1)将QC结果展示为小提琴图,features中的名称加引号,ncol=3表示图形分三列展示
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
进行质控后散点图(散点图)
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
if (!require(patchwork))install.packages("patchwork")
CombinePlots(plots = list(plot1, plot2))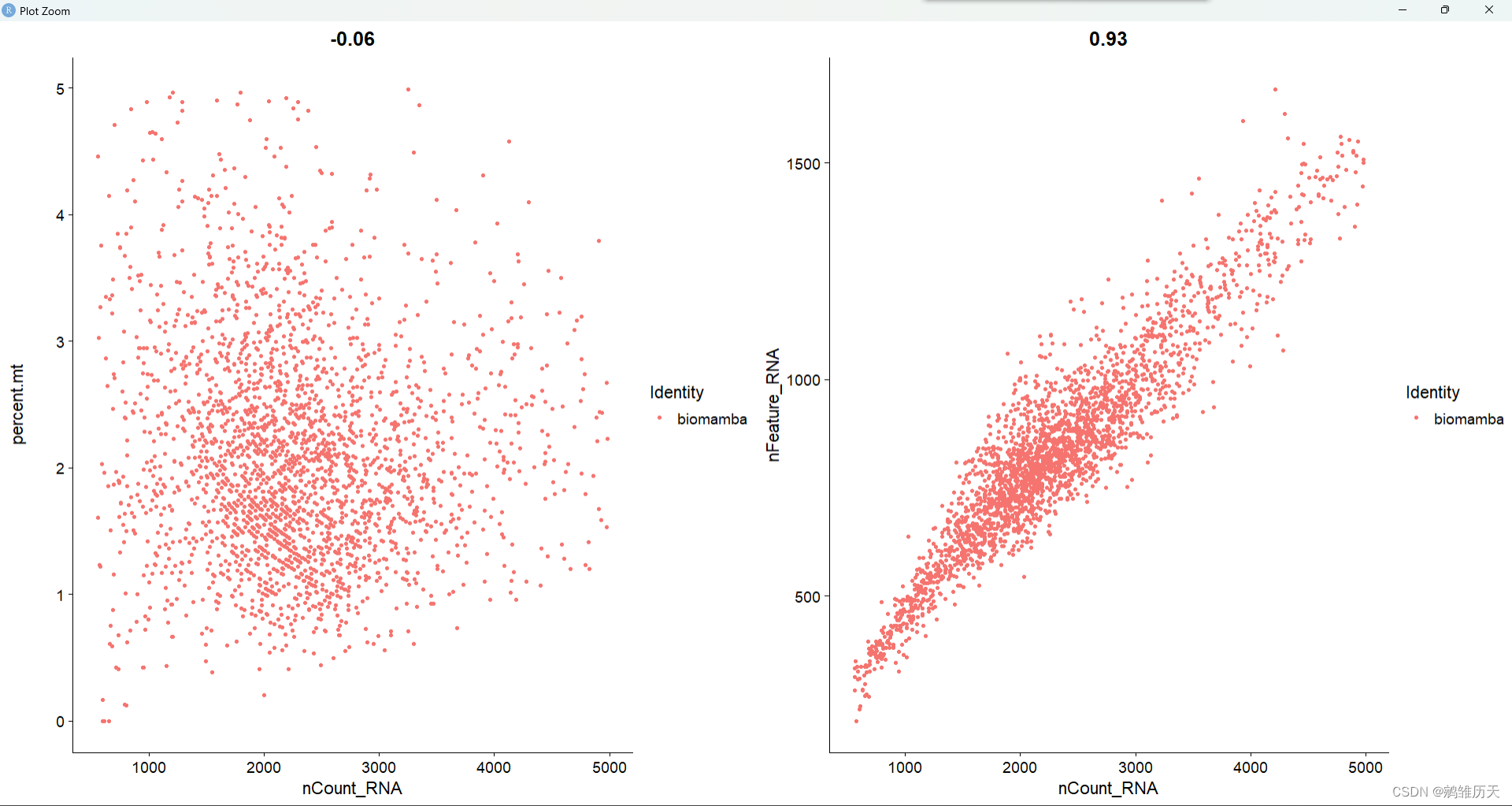
nCount(总基因表达数)、nFeature(总基因数)、percent.HB(红细胞基因表达⽐例)、percent.MT(线粒体基因表达⽐例)
数据标准化
方法一 normalization函数,默认LogNormalize的方法
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)方法二 SCTransform,是一种三合一的方法,可以将质控,归一化和去识别高变基因合为一体
pbmc <- SCTransform(pbmc, vars.to.regress = "percent.mt", verbose = FALSE)数据标准化的意义
- 去除测序深度带来的影响:
- 例:细胞A中总reads数10,基因a的reads数为2;细胞B中总reads数1000,基因a的reads数为20。并不能说基因a在细胞B中表达量大于A,故需要进行标准化。
- 标准化原则:
- 每个细胞的每个基因的count数除以该细胞总count数,然后乘以因子(10000),再进行log(n+1)转换
- 标准化后的数据保存在pbmc@assays
高变基因的选择
FindVariableFeatures()参数意义:
- FindVariableFeatures 函数有 3 种选择高表达变异基因的方法,可以通过 selection.method参数来选择,它们分别是: vst(默认值), mean.var.plot 和 dispersion。 nfeatures 参数的默认值是 2000,可以改变。如果 selection.method 参数选择的是 mean.var.plot,就不需要人为规定高表达变异基因的数目,算法会自动选择合适的数目。 建议使用完 FindVariableFeatures 函数后,用 VariableFeaturePlot 对这些高表达变异基因再做个可视化,看看使用默认值 2000 会不会有问题。
#选择高变基因,使用“vst”方法选择2000个高变基因
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
#储存前10位高变基因
top10 <- head(VariableFeatures(pbmc), 10)
plot3 <- VariableFeaturePlot(pbmc)#高变基因散点图
plot4 <- LabelPoints(plot=plot3,
points = top10,
repel=TRUE)#top10加上基因名标签
CombinePlots(plots = list(plot3, plot4), ncol =2)#结合到一张图中
寻找到的高变基因以及高变基因中的Top10
(二)PCA分析:线性降维
PCA分析,并且找到后续数据处理的维度
#数据标准化 Scale
#对所有基因进行标准化
# all.genes <- rownames(pbmc)
# pbmc <- ScaleData(pbmc, features = all.genes)
#只对上述2000个高变基因进行标准化,通常仅对高变基因进行标准化和降维
pbmc <- ScaleData(pbmc)
#PCA降维
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
#选择前5个维度进行查看
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
DimPlot(pbmc, reduction = "pca")
head(pbmc[['pca']]@cell.embeddings)[1:2,1:2]
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
#使用DimHeatmap可视化函数查看样品中前500个细胞在前6个PCA中的热图:
DimHeatmap(pbmc, dims = 1:6, cells = 500, balanced = TRUE)
#绘制ElbowPlot图
ElbowPlot(pbmc)
#绘制JackStraw图
pbmc <- JackStraw(pbmc,num.replicate = 100)
pbmc <- scoreJackStraw(pbmc,dims = 1:20)
JackStrawPlot(pbmc, dims = 1:20)
str(pbmc)ScaleData()标准化函数作用:
- 为后续PCA降维做准备。
- PCA降维要求数据为正态分布,即平均值为0,方差为1。
DimPlot()函数生成主成分分析结果图
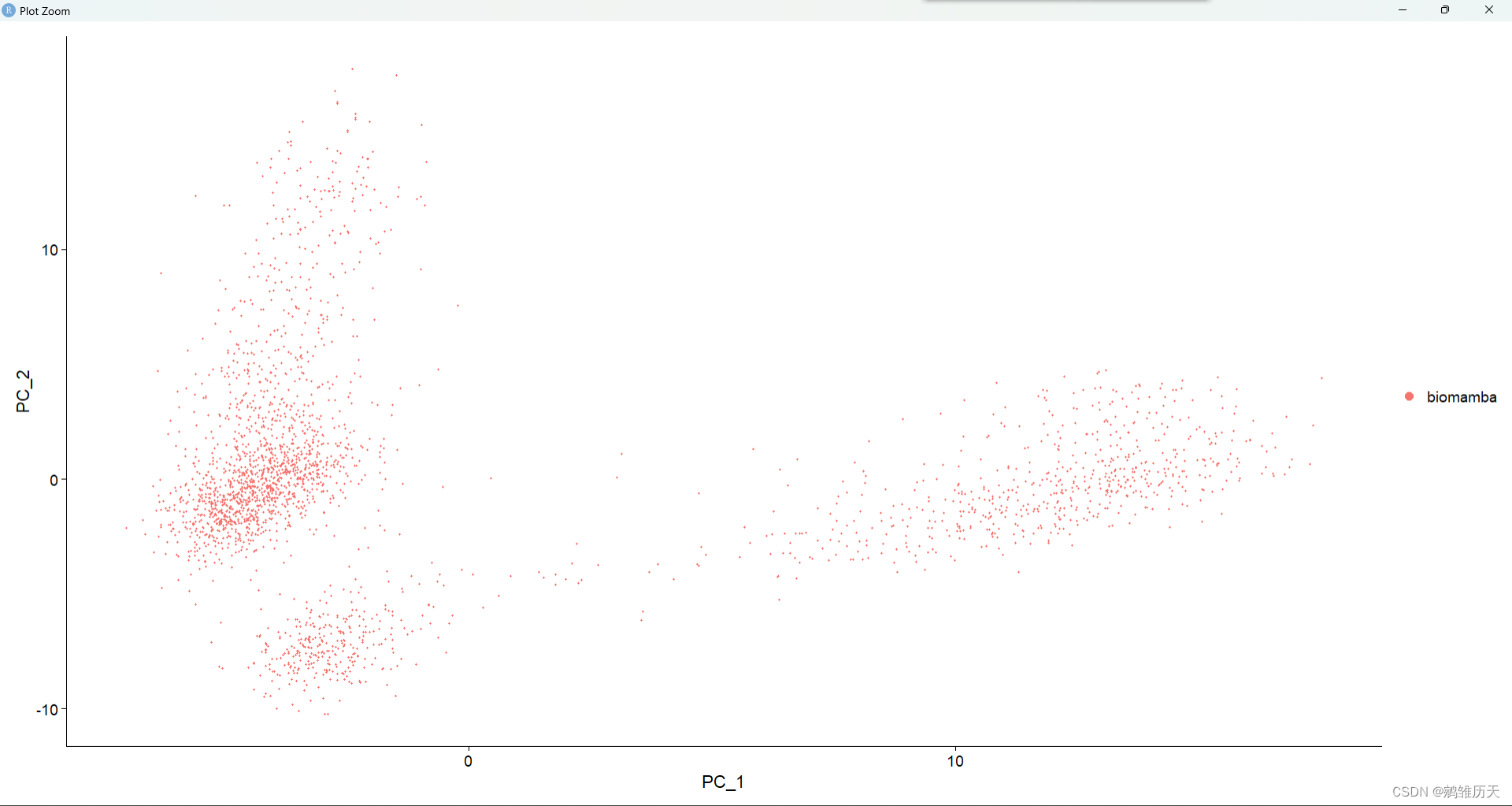
head(pbmc[['pca']]@cell.embeddings)[1:2,1:2]这段代码的含义是查看pbmc数据集中PCA降维后的细胞嵌入的前两行和前两列的数据。
输出结果如下:
PC_1 PC_2
AAACATACAACCAC-1 -4.8166626 0.3984358
AAACATTGAGCTAC-1 -0.6014593 -4.1511997这个结果表示PCA降维后的细胞嵌入中的前两行数据,其中第一列是PC_1的值,第二列是PC_2的值。每一行代表一个细胞,在低维空间中的位置由PC_1和PC_2的值表示。
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")

#使用DimHeatmap可视化函数查看样品中前500个细胞在前6个PCA中的热图:
DimHeatmap(pbmc, dims = 1:6, cells = 500, balanced = TRUE)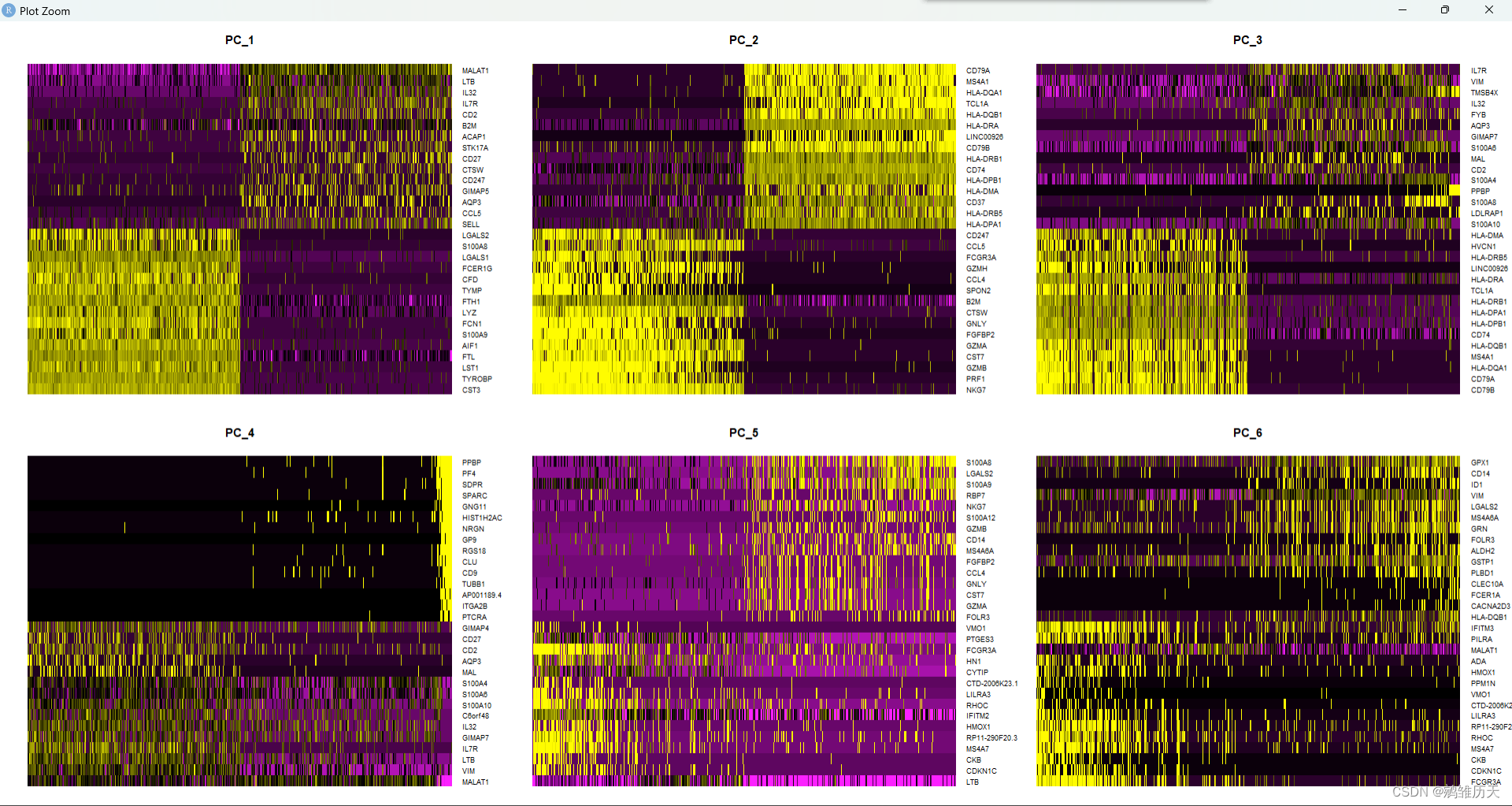
确定数据集的维度
目的:每个维度(pc)本质上代表一个“元特征”,它将相关特征集中的信息组合在一起。因此,越在顶部的主成分越可能代表数据集。然而,我们应该选择多少个主成分才认为我们选择的数据包含了绝大部分的原始数据信息呢?
方法
(1)ElbowPlot函数,基于每个主成分所解释的方差百分比的排序,通过寻找“拐点”来判断几个维度可包含数据的大部分信息。

(2)JackStraw()函数, 使用基于零分布的置换检验方法。JackStraw分析是一种常用的统计方法,用于评估单细胞RNA测序数据中的主要和次要成分是否具有统计显著性。它基于随机化方法,通过将原始数据中的样本标签进行随机重排,来估计每个主成分的p值。这样可以判断哪些主成分是真实的信号,而不是随机噪声。JackStrawPlot()函数提供可视化方法,用于比较每一个主成分的p-value的分布,虚线是均匀分布;显著的主成分富集有小p-Value基因,实线位于虚线左上方。
具体解释如下:
pbmc <- JackStraw(pbmc, num.replicate = 100)
#对pbmc数据集进行JackStraw分析。JackStraw函数用于计算主成分的p值和Q值
#num.replicate参数指定了随机重排的次数,这里设置为100次
pbmc <- scoreJackStraw(pbmc, dims = 1:20)
#使用scoreJackStraw函数对pbmc数据集进行评分
#dims参数指定了要评分的主成分的范围,这里选择了前20个主成分
JackStrawPlot(pbmc, dims = 1:20)
#使用JackStrawPlot函数绘制JackStraw图
#JackStraw图以p值和Q值作为纵轴,主成分的编号作为横轴,展示主成分的显著性和噪声水平通过JackStraw分析和JackStraw图,我们可以评估每个主成分的显著性,并确定哪些主成分是真实的信号,进而进行后续的数据解释和分析。

(三)细胞聚类
将具有相似基因表达模式的细胞之间绘制边缘,然后将他们划分为一个内联群体
并进行tSNE和UMAP分析
注意:umap的使用需要先在电脑上搭建python环境,检查是否下载成功标志就是:library(umap)能否成功
# 细胞聚类
# tSNE聚类与UMAP聚类选择一个执行即可,都是基于PCA降维的结果进行聚类
# 此处维度选择10是根据上述“ElbowPlot(pbmc)”生成的主成分图确定的,图中拐点为10,表示10个维度足以反映高维信息
#计算最邻近距离
pbmc <- FindNeighbors(pbmc, dims = 1:10) # Louvain cluster graph based
#聚类,包含设置下游聚类的“间隔尺度”的分辨率参数resolution ,增加值会导致更多的聚类。
pbmc <- FindClusters(pbmc, resolution = 0.5)
#可以使用idents函数找到聚类情况:
#查看前5个细胞的聚类id
head(Idents(pbmc), 5)
#UMAP降维
pbmc <- RunUMAP(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "umap")
#tSNE降维
#pbmc <- RunTSNE(pbmc, dims = 1:10)
#DimPlot(pbmc, reduction = "tsne")UMAP降维聚类
#查看FCGR3A和CD14基因分布
FeaturePlot(pbmc,features = c('FCGR3A','CD14'),reduction = 'umap')

TSNE降维聚类

t-SNE vs UMAP
t-SNE(t-Distributed Stochastic Neighbor Embedding)和UMAP(Uniform Manifold Approximation and Projection)都是常用的非线性降维方法,用于将高维数据映射到低维空间。以下是它们的一些优劣比较:
t-SNE的优势:保留局部结构:t-SNE能够更好地保留数据的局部结构,尤其适用于可视化和聚类分析。它可以帮助发现数据中的类别、簇或群集,并在降维后的空间中保持它们的相对距离。
t-SNE的劣势:计算复杂度高:t-SNE的计算复杂度较高,尤其是在大规模数据集上。它的运行时间和内存消耗通常比较大,可能需要更多的计算资源和时间。tSNE的时间复杂度为O(n2),UMAP的时间复杂度为O(n1.14)
UMAP的优势:保留全局结构:UMAP相对于t-SNE来说更擅长保留数据的全局结构。它能够更好地捕捉数据之间的整体关系和距离,对于发现数据集中的全局模式和结构更有优势,更好的保留真实距离信息
UMAP的劣势:参数选择:UMAP有一些参数需要调整,如邻域大小、最小距离等。这些参数的选择可能对最终的降维结果产生影响,需要一定的经验和实验来确定最佳的参数设置。
综上所述,选择t-SNE还是UMAP取决于具体的数据集和分析目的。如果重点是保留局部结构和发现数据中的类别或簇,则t-SNE可能更适合。如果需要保留全局结构和捕捉数据之间的整体关系,则UMAP可能更适合。在实际应用中,可以尝试使用两种方法,并比较它们在降维效果和计算效率上的差异,选择最适合的方法。
【干货】高分SCI要用tSNE还是UMAP? - 知乎 (zhihu.com)
#添加细胞类群的标签
DimPlot(pbmc, reduction = "umap",label = TRUE)
LabelClusters(DimPlot(pbmc, reduction = "umap"),id = 'ident')
#可以在这里保存为以下形式,就不必再执行以上步骤
saveRDS(pbmc, file = "../output/pbmc_tutorial.rds")(四)差异分析:寻找marker gene
通过差异表达找到每个聚类的marker gene,差异分析可以有多种形式,如找到所有聚类的marker gene(如cluster1中所有的markgene是指cluster1相对于其余所有cluster是差异的)、两个cluster之间的差异分析、某个cluster中两个样品之间差异分析等
#差异分析
# find all markers of cluster 0(寻找cluster0的基因)
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, min.pct = 0.25)
head(cluster0.markers, n = 5)
#可以根据需要,将每个聚类的差异基因都找一遍,方便后续分析
#head(cluster0.markers, n = 5),可以将n = 5进行调整,比如换成10,即可显示10个差异基因
###参数解释
#ident.1 设置待分析的细胞类别
#min.pct 在两组细胞中的任何一组中检测到的最小百分
#thresh.test 在两组细胞间以一定数量的差异表达(平均)
#max.cells.per.ident 通过降低每个类的采样值,提高计算速度
#寻找cluster1的差异基因
cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)
#找到cluster5和cluster0、3之间的markgene
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
#找到每个cluster相比于其余cluster的markgene,只报道阳性的markgene
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
## 所有基因先分组,再根据avg_log2FC进行排序
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)FindAllMarkers()参数意义:
- only.pos = TRUE:只寻找上调的基因
- min.pct = 0.1:某基因在细胞中表达的细胞数占相应cluster细胞数最低10%
- logfc.threshold = 0.25 :fold change倍数为0.25
- 该函数是决速步,执行比较耗时。
根据avg_log2FC进行排序

#VlnPlot: 基于细胞类群的基因表达概率分布
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)

(五)可视化标记基因,即细胞注释
秀儿!10+生信分析最大的难点在这里!30多种方法怎么选?今天帮你解决!_pbmc (sohu.com)
#对marker_gene进行筛选p_val<0.05
pbmc.markers %>% subset(p_val<0.05)
## 所有基因先分组,再根据avg_log2FC进行排序,选出每组前十个
list_marker <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
print(list_marker,n = Inf)#打印list_marker所有值
#保存差异分析结果到csv
df_marker=data.frame(p_val = list_marker$p_val,
avg_log2FC = list_marker$avg_log2FC,
pct.1 = list_marker$pct.1,
pct.2 = list_marker$pct.2,
p_val_adj = list_marker$p_val_adj,
cluster = list_marker$cluster,
gene = list_marker$gene)
write.csv(df_marker,"marker.csv")
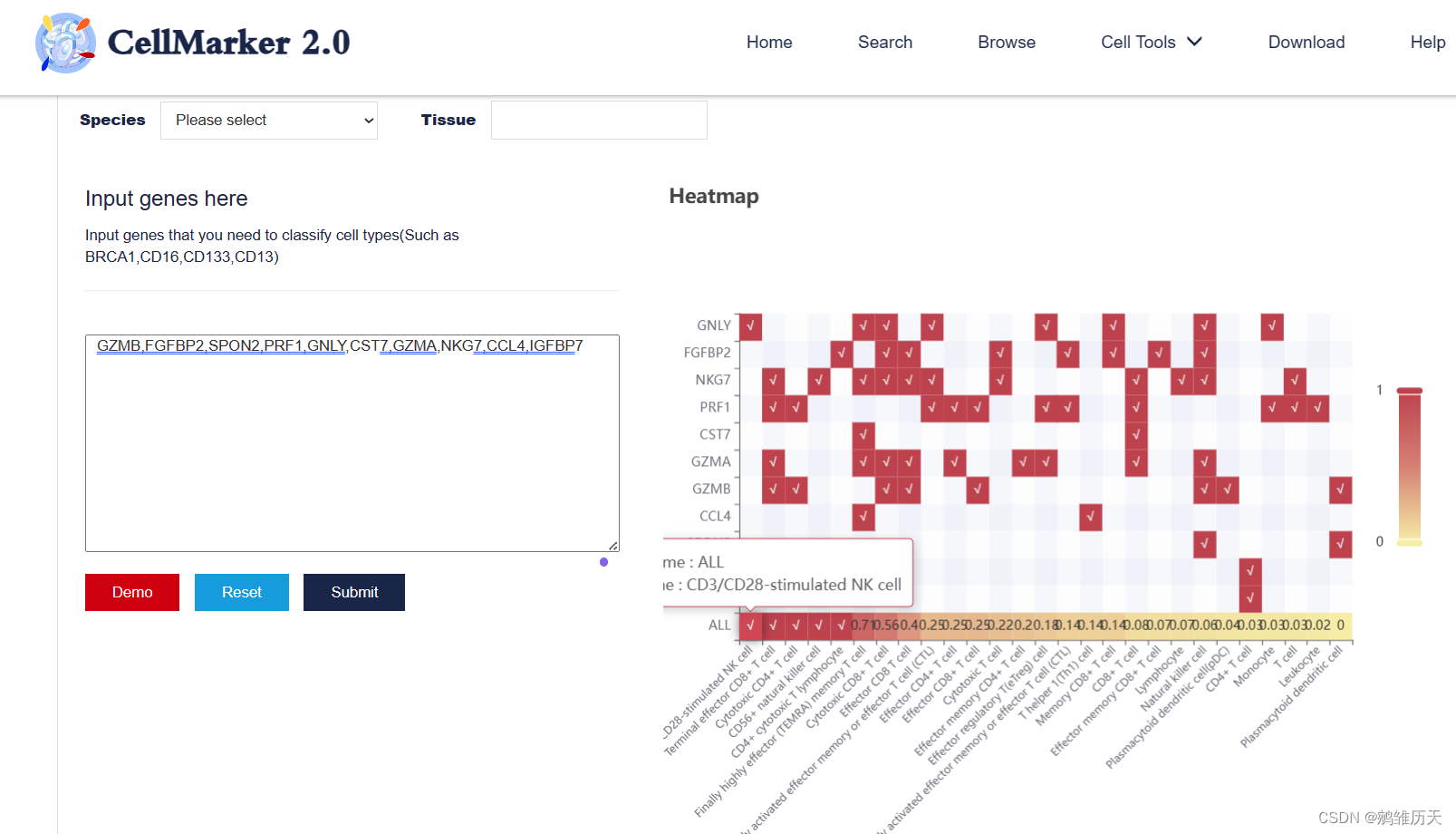
#添加细胞注释信息,通过CellMarker注释每一个cluster代表的细胞类群
new.cluster.ids <- c("Memory CD4 T", "Activated effector", "Pro-inflammatory macrophage", "CDC2", "CD8 T", "Non-classical monocyte",
"CD3/CD28-stimulated NK", "Megakaryocyte")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()将csv文件中各组的top10marker基因数据处理成便于CellMarker需求形式

使用CellMarker中的cell annotation工具进行注释CellMarker2.0 (hrbmu.edu.cn)
展示注释后的DimPlot图

小白第一次写文章,欢迎大家评论区指正!