GuacaMol: Benchmarking Models for de Novo Molecular Design【2019】
GuacaMol:新分子设计的基准模型
paper:https://pubs.acs.org/doi/10.1021/acs.jcim.8b00839
接口 code:https://github.com/BenevolentAI/guacamol
Benchmark code:https://github.com/benevolentAI/guacamol_baselines
一、摘要:
De novo design seeks to generate molecules with required property profiles by virtual design-make-test cycles.
With the emergence of deep learning and neural generative models in many application areas, models for molecular design based on neural networks appeared recently and show promising results. However, the new models have not been profiled on consistent tasks, and comparative studies to well established algorithms have only seldom been performed. To standardize the assessment of both classical and neural models for de novo molecular design, we propose an evaluation framework, GuacaMol, based on a suite of standardized benchmarks. The benchmark tasks encompass measuring the fidelity of the models to reproduce the property distribution of the training sets, the ability to generate novel molecules, the exploration and exploitation of chemical space, and a variety of single and multiobjective optimization tasks.
二、 评估新设计的技术
To profile(描述) models for de novo molecular design, we differentiate between their two main use cases:
- Given a training set of molecules, a model generates new molecules following the same chemical distribution.【Distribution-Learning Benchmarks】
- A model generates the best possible molecules to satisfy a predefined goal.【Goal-Directed Benchmarks.】
The collection of benchmarks we propose below assesses both facets defined here. In the following we will refer to these two categories as distribution-learning benchmarks and goal-directed benchmarks, respectively.
3.1). Distribution-Learning Benchmarks.【分布学习的基准】
分布学习的基准是:评估模型学习生成类似于训练集的分子的效果,在这项工作中,训练集是ChEMBL。
The distribution-learning benchmarks assess how well models learn to generate molecules similar to a training set, which in this work is a standardized subset of the ChEMBL database (see Appendix 8.1). We consider five benchmarks for distribution learning:
3.1.1. Validity(有效性).
Validity就是看生成的分子的SMILES是否是理性的。
The validity benchmark assesses whether the generated molecules are actually valid, that is, whether they correspond to a (at least theoretically) realistic molecule. For example, molecules with an incorrect SMILES syntax, or with invalid valence, are penalized.
3.1.2. Uniqueness(独一无二性).
是否总是生成一个分子多次?
Given the high dimensionality of chemical space and the huge number of molecules potentially relevant in medicine, generative models should be able to generate a vast number of different molecules. The uniqueness benchmark assesses whether models are able to generate unique molecules--that is, if a model generates the same molecule multiple times, it will be penalized.
3.1.3. Novelty(新颖性).
模型是否生成存在于训练集中的分子?
Since the ChEMBL training set represents only a tiny subset of drug-like chemical space, a model for de novo molecular design with a good coverage of chemical space will rarely generate molecules present in the training set. The novelty benchmark penalizes models when they generate molecules already present in the training set. Therefore, models overfitting the training set will obtain low scores on this task.
3.1.4. Frechet ChemNet Distance.(FCD)
来源于:Fréchet ChemNet Distance: A metric for generative models for molecules in drug discovery(https://arxiv.org/abs/1803.09518)
评估生成的分子与训练集中的分子之间的相似性,越相似,FCD分数越低。
Preuer et al. introduced the Frechet ChemNet Distance (FCD) as a measure of how close distributions of generated data are to the distribution of molecules in the training set. In the context of drug discovery, the usefulness of this metric stems from its ability to incorporate important chemical and biological features. The FCD is determined from the hidden representation of molecules in a neural network called ChemNet trained for predicting biological activities, similarly to the Frechet Inception Distance(FID) sometimes applied to generative models for images. More precisely, the means and covariances of the activations of the penultimate layer of ChemNet are calculated for the reference set and for the set of generated molecules. The FCD is then calculated as the Frechet ́ distance for both pairs of values. Similar molecule distributions are characterized by low FCD values.
3.1.5. KL Divergence(KL 散度).
衡量分布P与分布Q近似程度。
The Kullback−Leibler (KL) divergence measures how well a probability distribution Q approximates another distribution P. For the KL divergence benchmark, the probability distributions of a variety of physicochemical descriptors for the training set and a set of generated molecules are compared, and the corresponding KL divergences are calculated. Models able to capture the distributions of molecules in the training set will lead to small KL divergence values.
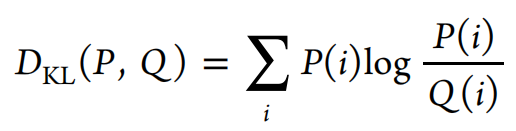
3.2). Goal-Directed Benchmarks.【目标导向的基准】
3.2.1. Similarity(相似性).
Similarity is one of the core concepts of chemoinformatics.
3.2.2. Rediscovery(重新发现).
Rediscovery benchmarks are closely related to the similarity benchmarks described above.
3.2.3. Isomers.(同分异构体)
For the isomer benchmarks, the task is to generate molecules that correspond to a target molecular formula (e.g., ).
3.2.4. Median Molecules.(中间分子)
In the median molecules benchmarks, the similarity to several molecules must be maximized simultaneously; it is designed to be a conflicting task.
4. METHODS
GuacaMol提供了用户友好的接口,允许研究人员以最小的代价将他们的模型与基准测试框架相结合:
Our framework for benchmarking models for de novo design, GuacaMol, is available as an open-source Python package and can be downloaded from the Internet at: www.github.com/BenevolentAI/guacamol. GuacaMol provides user-friendly interfaces that allow researchers to couple their models to the benchmarking framework with minimal effort.
We selected five distribution-learning and 20 goal-directed benchmarks. The exhaustive specifications and implementation details of the distribution-learning and goal-directed benchmarks are given in Appendices 7.2 and 7.3, respectively.
For all chemoinformatics-related operations, such as handling or canonicalization of SMILES strings, physicochemical property (logP, TPSA), or similarity calculations, GuacaMol relies on the RDKit package.
Several of the molecule generation approaches we studied require a training data set. ChEMBL was used for this purpose. The generation of the data set is discussed and detailed in Appendix 7.1.
除了引入benchmark,这个模型还提供了在多个baseline下的评估,目的是公平的评估SOTA方法。
In addition to the introduction of a benchmark suite for generative chemistry, this paper provides its evaluation over several baselines. The goal is to present a fair comparison of some recent methods that seem to achieve state-of-the-art results in the field. These have been selected to represent a variety of methods, ranging from deep learning generative models to established genetic algorithms and more recent Monte Carlo Tree Search (MCTS). Internal molecule representation was also taken into account, so that methods using SMILES strings were represented alongside others using a graph representation of atoms and bonds. For completeness and to provide a lower bound on the benchmark scores, two dummy baselines, performing random sampling(Distribution-learning Benchmark) and best of data set picking(Goal-direction Benchmark), are also provided. All the baseline models assessed in this work are described in Appendix 7.4. Our implementation of those models is available on GitHub.(GitHub - BenevolentAI/guacamol_baselines: Baselines models for GuacaMol benchmarks)
5. RESULTS AND DISCUSSION
5.1. Distribution-Learning Benchmarks.
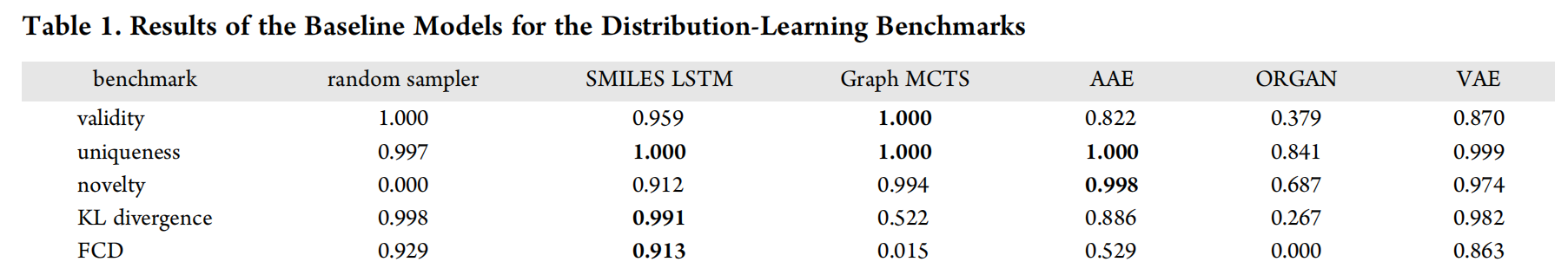
Table 1 lists the results for the distribution-learning benchmarks.
- The random sampler model is a useful baseline for comparison, because it shows what scores can be expected for good models on the KL divergence and Frechet benchmarks. Unsurprisingly, its novelty score is zero, since all the molecules it generates are present in the data set.(random sample是在数据集中取出的,它的作用是作为baseline进行比较)
- The SMILES LSTM model sometimes produces invalid molecules. However, they are diverse, as attested by the uniqueness and novelty benchmarks, and closely resemble molecules from ChEMBL.
- The Graph MCTS model is characterized by a high degree of valid, unique, and novel molecules. Nevertheless, it is not able to reproduce the property distributions of the underlying training set as precisely as the SMILES LSTM model.
- The AAE model obtains excellent uniqueness and novelty scores. The molecules this model produces are, however, not as close to the data set as the ones generated by the SMILES LSTM model.
- ORGAN obtains poor scores for all the distribution-learning tasks. More than half the molecules it generates are invalid, and they do not resemble the training set, as shown by the KL divergence and FCD scores. This might indicate mode collapse, which is a phenomenon often observed when training GANs.
- The scores of the VAE model are not the best in any categories, but this model consistently produced relatively good scores for all the tasks.
Datasets :SMILES strings and their depictions for molecules generated by all six baseline models are available in the Supporting Information or on https://benevolent.ai/guacamol.
7.2. Implementation Details: Distribution-Learning Benchmarks
(1)Validity.
在10k个生成的分子中看能被RDKit成功解释的分子比例。
The validity score is calculated as the ratio of valid molecules out of 10 000 generated molecules. Molecules are considered to be valid if their SMILES representation can be successfully parsed by RDKit.
(2)Uniqueness.
10000个不同(不重复)的分子SMILES / 10000个分子
To calculate the uniqueness score, molecules are sampled from the generative model, until 10 000 valid molecules have been obtained. Duplicate molecules are identified by identical canonical SMILES strings. The score is obtained as the number of different canonical SMILES strings divided by 10 000.
(3)Novelty.
生成的分子中不在ChEMBL数据库中的比例
The novelty score is calculated by generating molecules, until 10 000 different canonical SMILES strings are obtained, and computing the ratio of molecules not present in the ChEMBL data set.
(4)Frechet ChemNet Distance (FCD).
从ChEMBL数据库中取出10000个分子,然后随机取样10000个生成的分子,计算它们之间的相似度。
To generate the FCD score, a random subset of 10 000 molecules is selected from the ChEMBL data set, and the generative model is sampled, until 10 000 valid molecules are obtained. The FCD between both sets of molecules is calculated with the FCD package available on GitHub【FCD Package. https://github.com/bioinf-jku/FCD】, and the final score S is given by
![]()
(5)KL Divergence.
使用RDKit对sample sets和reference sets计算以下物理化学描述符:
For this task, the following physicochemical descriptors are calculated with RDKit for both the sampled and the reference sets: BertzCT, MolLogP, MolWt, TPSA, NumHAcceptors, NumHDonors, NumRotatableBonds, NumAliphaticRings, and NumAromaticRings. Furthermore, we calculate the distribution of maximum nearest neighbor similarities on ECFP4 fingerprints for both sets. Then, the distribution of these descriptors is computed via kernel density estimation (using the scipy package) for continuous descriptors or as a histogram for discrete descriptors. The KL divergence DKL,i is then computed for each descriptor i and is aggregated to a final score S via
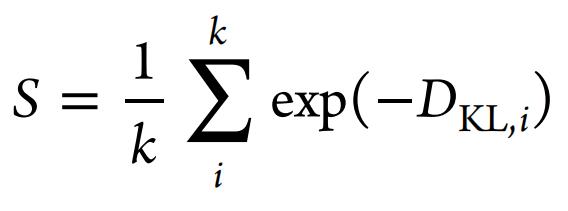
where k is the number of descriptors (in our case k = 9).