Resumen
Dado que el tutorial es relativamente largo, no se recomienda practicarlo, simplemente léalo para comprenderlo y aprender.
Antes de ejecutar un análisis filogenético basado en la probabilidad, el usuario debe decidir qué parámetros libres deben incluirse en el modelo: si se debe asumir una tasa única para todas las sustituciones (como el modelo Jukes-Cantor de evolución de secuencias) o si se deben asumir diferentes tasas de transición. y se deben permitir interrupciones Tipo de cambio (como el modelo HKY). ¿O deberían usarse diferentes proporciones para todas las sustituciones (como en el modelo GTR), deberían estimarse o asumirse que las frecuencias de los cuatro nucleótidos ("frecuencias de estado") son todas iguales? El número óptimo de parámetros libres del modelo depende de los datos disponibles y se puede elegir en función de criterios como el Criterio de Información de Akaike (AIC), que busca equilibrar la mejora en el ajuste del modelo con el número de parámetros adicionales necesarios para el ajuste del modelo.
En este tutorial [1] describiré cómo seleccionar modelos alternativos para el análisis filogenético utilizando el software PAUP* (Swofford 2003), una herramienta popular y versátil para varios tipos de análisis filogenéticos.
conjunto de datos
Los datos utilizados en este tutorial son una versión filtrada de la alineación generada para las secuencias 16s y RAG1 en el tutorial de alineación de secuencias múltiples. Dado que PAUP* requiere la alineación del formato Nexus como entrada, utilice los archivos 16s_filtered.nex y rag1_filtered.nex.
PAÚA*
Desarrollado originalmente a finales de la década de 1980, este software es uno de los programas de análisis filogenético más antiguos y, aunque existe desde hace mucho tiempo, su autor Dave Swofford nunca lanzó una versión final. Aunque PAUP* ha sido superado durante mucho tiempo en velocidad por otros programas de inferencia filogenética basada en probabilidades, sigue siendo importante por la variedad de otras funciones que contiene. No hace mucho, PAUP* sólo se podía comprar en Sinauer Associates por unos 100 dólares. Desde 2015, Dave Swofford ha estado distribuyendo la versión actualizada de PAUP* 4.0 de forma gratuita como versión de prueba en su nuevo sitio web PAUP*. Estas pruebas caducan después de unos meses, por lo que es posible que tengas que descargarlas nuevamente si deseas utilizar PAUP en el futuro. Esta situación puede ser sólo temporal, ya que el desarrollo de PAUP 5 está en curso y el producto se distribuirá comercialmente, al menos parcialmente.
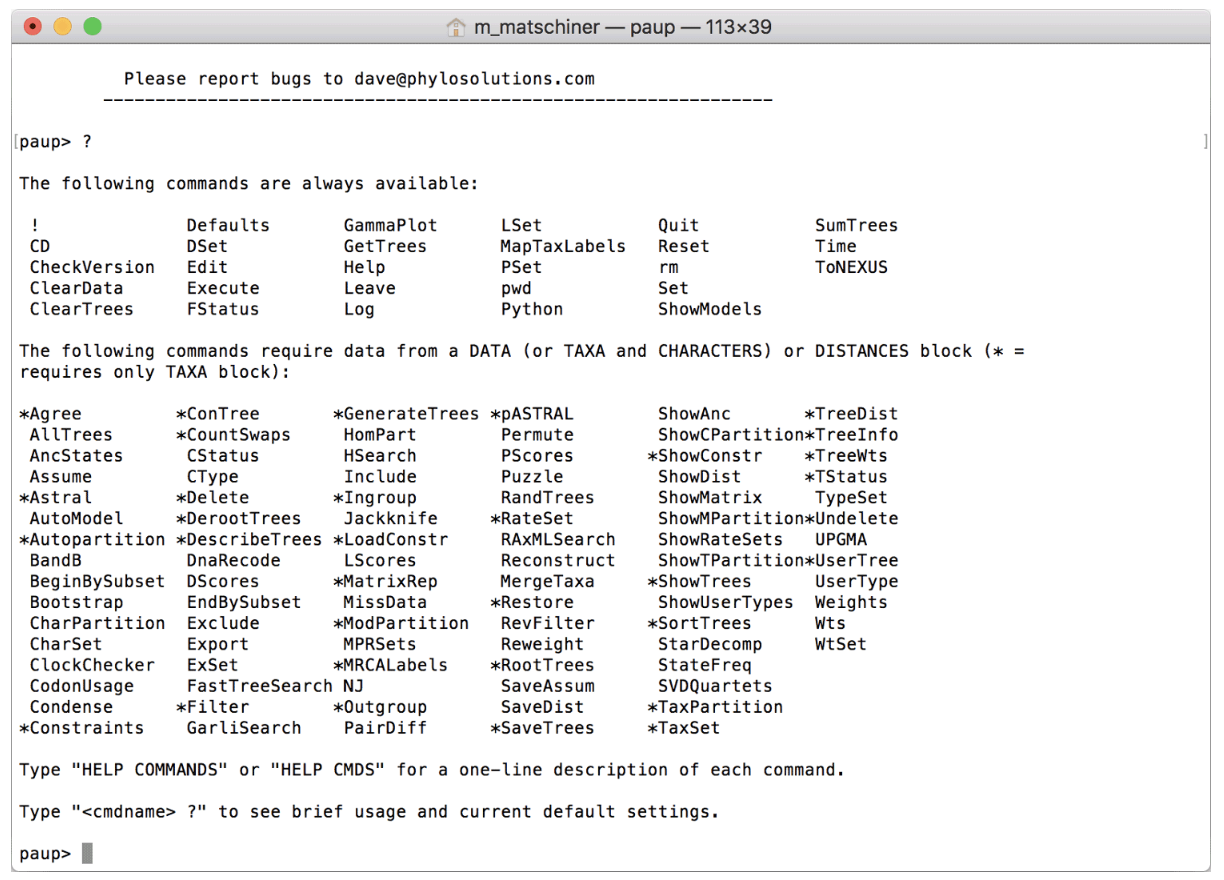
Aunque las descripciones de este tutorial suponen que ha instalado la versión de interfaz gráfica de usuario (GUI) de PAUP* para Mac OS X o Windows, también puede instalar la versión de línea de comandos de PAUP*, que se requiere en Linux o Mac OS X. Catalina o posterior, ya que actualmente no existe una GUI para estos sistemas. Si está utilizando la versión de línea de comandos, es posible que necesite buscar el comando equivalente; esto siempre se puede hacer a través de la pantalla de ayuda de PAUP* después de iniciar PAUP*, que se puede mostrar escribiendo "?". y presione Entrar. La siguiente captura de pantalla muestra la pantalla de ayuda para la versión de línea de comando de PAUP*.
Selección de modelos e inferencia filogenética.
Las comparaciones del ajuste de modelos basados en sustitución con datos de secuencia se han implementado en una variedad de herramientas y se realizan más comúnmente utilizando el programa jModelTest. Pero dado que la selección automática de modelos alternativos se implementó recientemente en PAUP*, y los otros tutoriales en este repositorio requieren que PAUP esté instalado de todos modos , estoy usando PAUP aquí en lugar de jModelTest para la selección de modelos. De hecho, la selección de modelos entre los dos programas es muy similar.
-
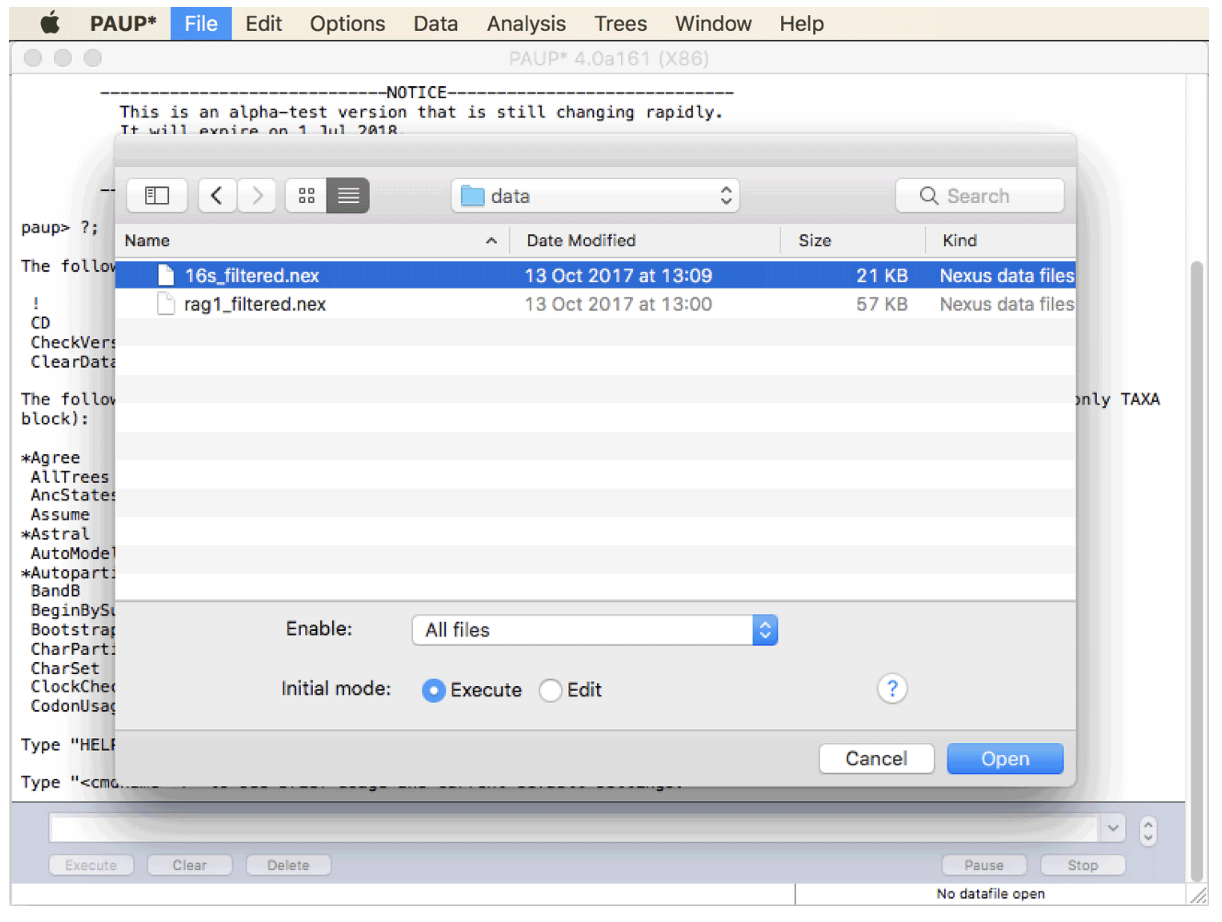
单击 PAUP* 的“文件”菜单中的“打开...”。确保在打开的窗口底部选择“执行”作为初始模式,如下一个屏幕截图所示。选择 Nexus 格式的 16s 序列对齐文件 (16s_filtered.nex),然后单击“打开”。 PAUP* 将给出其对该文件的解释的简短报告,包括在比对中发现的物种(分类单元)和字符的数量。
-
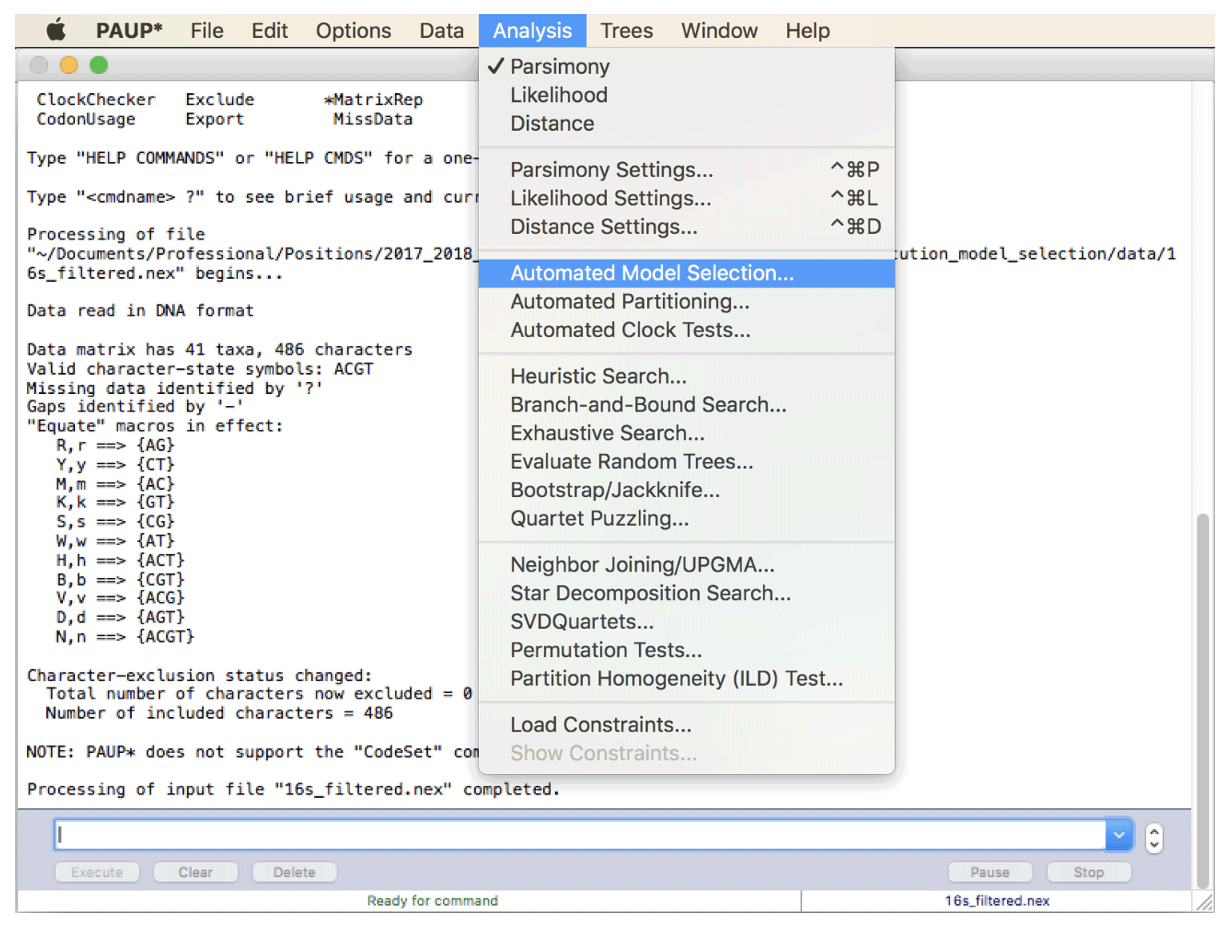
“自动模型选择”选项可以在 PAUP* 的“分析”菜单中找到。但是,当您单击它时,您会看到为了运行此模型选择,需要系统发育。虽然这可能看起来可能会导致循环推理(选择替代模型是最大似然系统发育分析所必需的,但也取决于系统发育),但这在实践中不是问题,因为模型选择的结果并不强烈依赖于正确的系统发育;因此,任何合理的系统发育都会导致相似的模型选择结果。因此,最好的解决方案是使用 Neighbor-Joining 算法运行快速系统发育分析,该算法也可以在 PAUP* 中方便地实现。
-
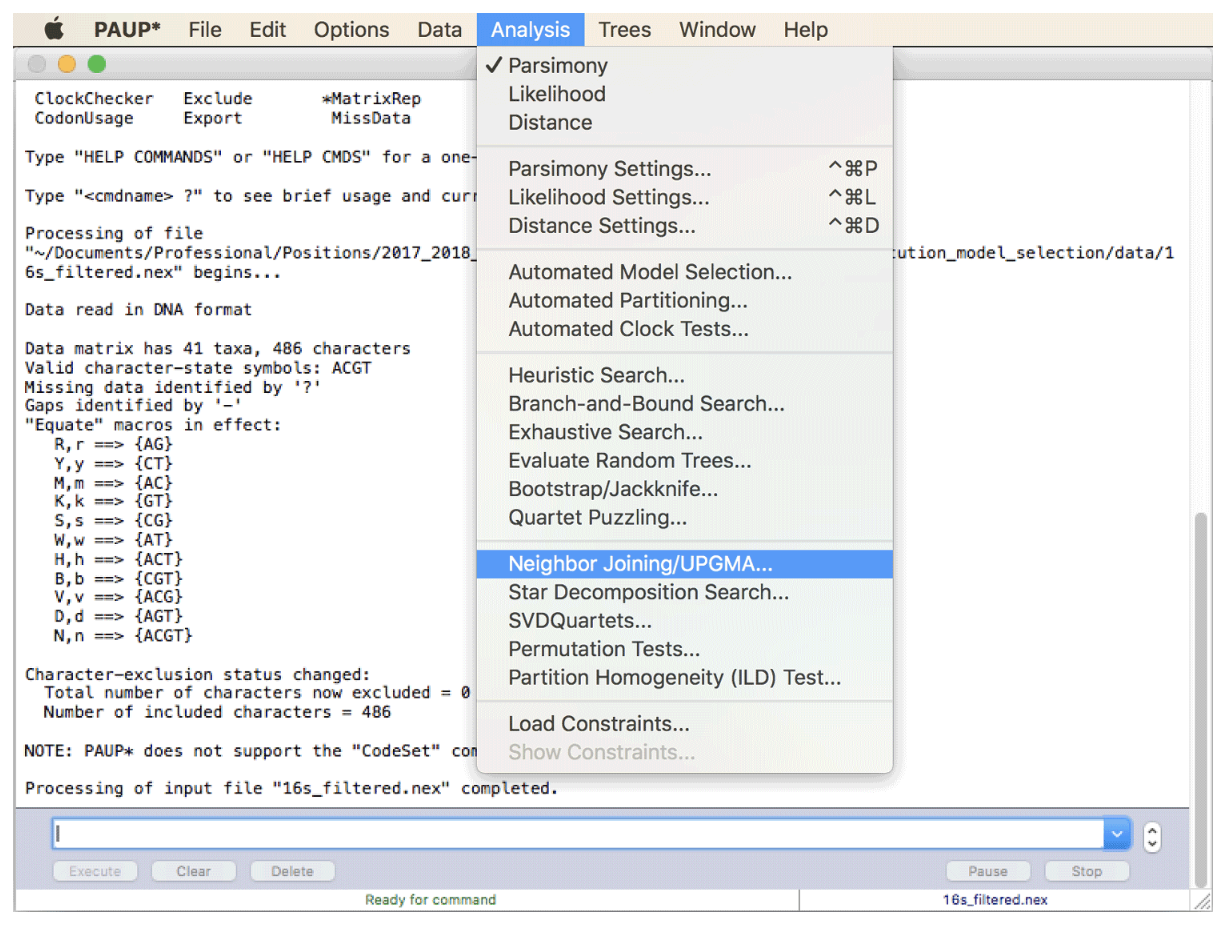
要从邻接系统发育分析的可用设置中进行选择,请单击 PAUP* 的“分析”菜单中的“邻接/UPGMA...”,如下面的屏幕截图所示。
-
在新打开的弹出窗口中,保留所有默认选项并单击“确定”(PAUP* 命令行版本中的等效命令只是 NJ;)。 -
再次单击“分析”菜单中的“自动模型选择...”。使用邻接生成的树将已被选择用于模型选择,弹出窗口现在将为您提供用于此模型选择的多个选项。模型选择的可用标准称为“AIC”、“AICc”、“BIC”和“DT”。这些与似然比检验类似,但优点是它们可用于比较非“嵌套”模型(如果其中一个模型具有其他模型的所有参数加上附加参数,则两个模型是嵌套的)。 “AIC”代表“Akaike信息准则”,“AICc”是“针对小样本量校正的Akaike信息准则”,“BIC”是“贝叶斯信息准则”,“DT”是“决策理论”标准。其中最常用的是 Akaike 信息准则。每个模型的 AIC 独立计算为 AIC = 2 k −2 log(L),其中 k 是模型中自由参数的数量,L 是所有自由参数优化后数据的可能性(即最大可能性)。通常,如果一个模型的 AIC 分数比另一个模型的 AIC 分数好(= 小)至少 4 分,则该模型被认为优于另一个模型。设置“AIC”旁边的勾号,但删除“AICc”、“BIC”和“DT”旁边的勾号。另请选择“应用选择模型的设置:”右侧的“AIC”。作为“模型集”,选择数字“3”。这意味着将测试具有相等替代率的模型(例如 Jukes-Cantor 模型)、具有单独的转换和颠换替代率的模型(例如 HKY 模型)以及具有六个独立替代率的模型(GTR 模型)。保留“等速率”和“gamma”旁边的勾号(允许站点间速率变化的伽玛分布),但删除“invar.sites”和“两者”的勾号。我建议这样做,因为不变位点比例(“+I”)和位点间速率变化(“+G”)的参数很混乱,因为对一组位点应用特别低的速率几乎具有相同的效果。考虑到这些站点的效果完全不变。保留“显示每个模型的输出”旁边的勾号,并设置“显示每个模型的参数估计”旁边的勾号。确保设置面板如下面的屏幕截图所示,然后单击“确定”。
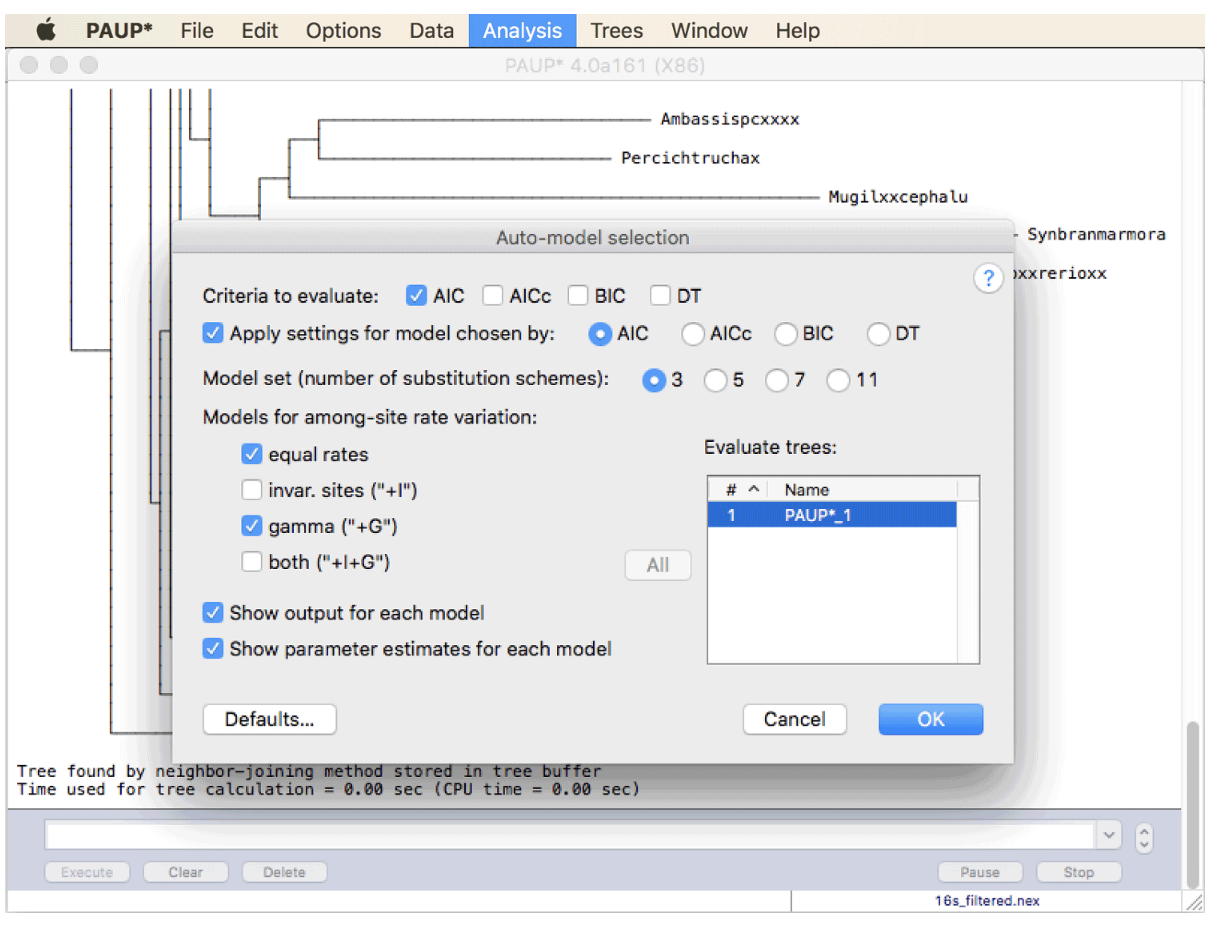
-
PAUP* 将在三个表中报告模型选择的输出。在第一个部分(在“评估树 1 的模型”下),您将看到已比较的 12 个模型的列表,如下所示(“JC”代表 Jukes-Cantor 模型)。
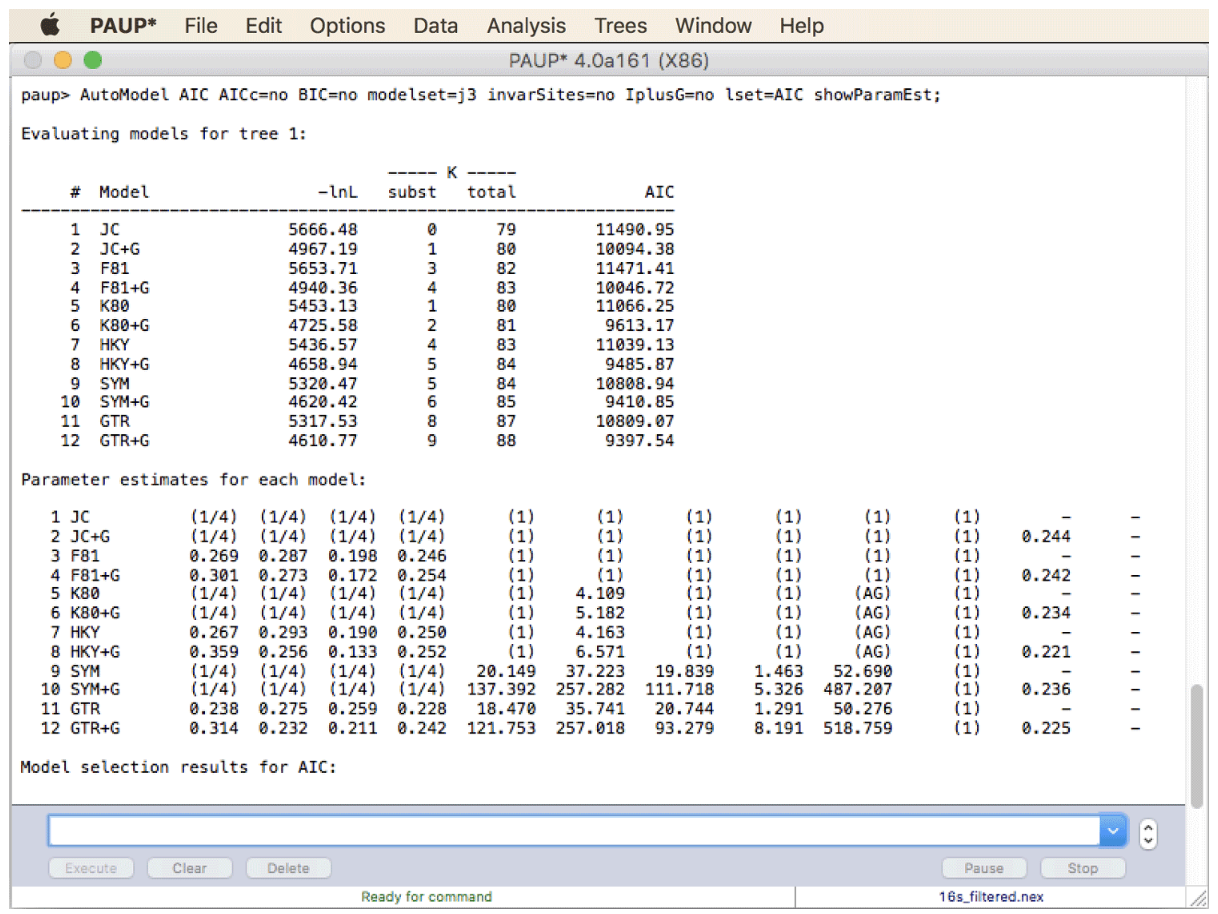
-
在同一个表的第 4 列和第 5 列中,您将看到 k,即模型中自由参数的数量。第 4 列列出了与最简单模型相比额外的自由参数的数量,第 5 列列出了自由参数的总数。第二个表列出了每个模型的参数估计值。每个型号的编号和名称后面有九列数字。最后,第三个表再次列出了模型,但这次是按 AIC 分数排名。
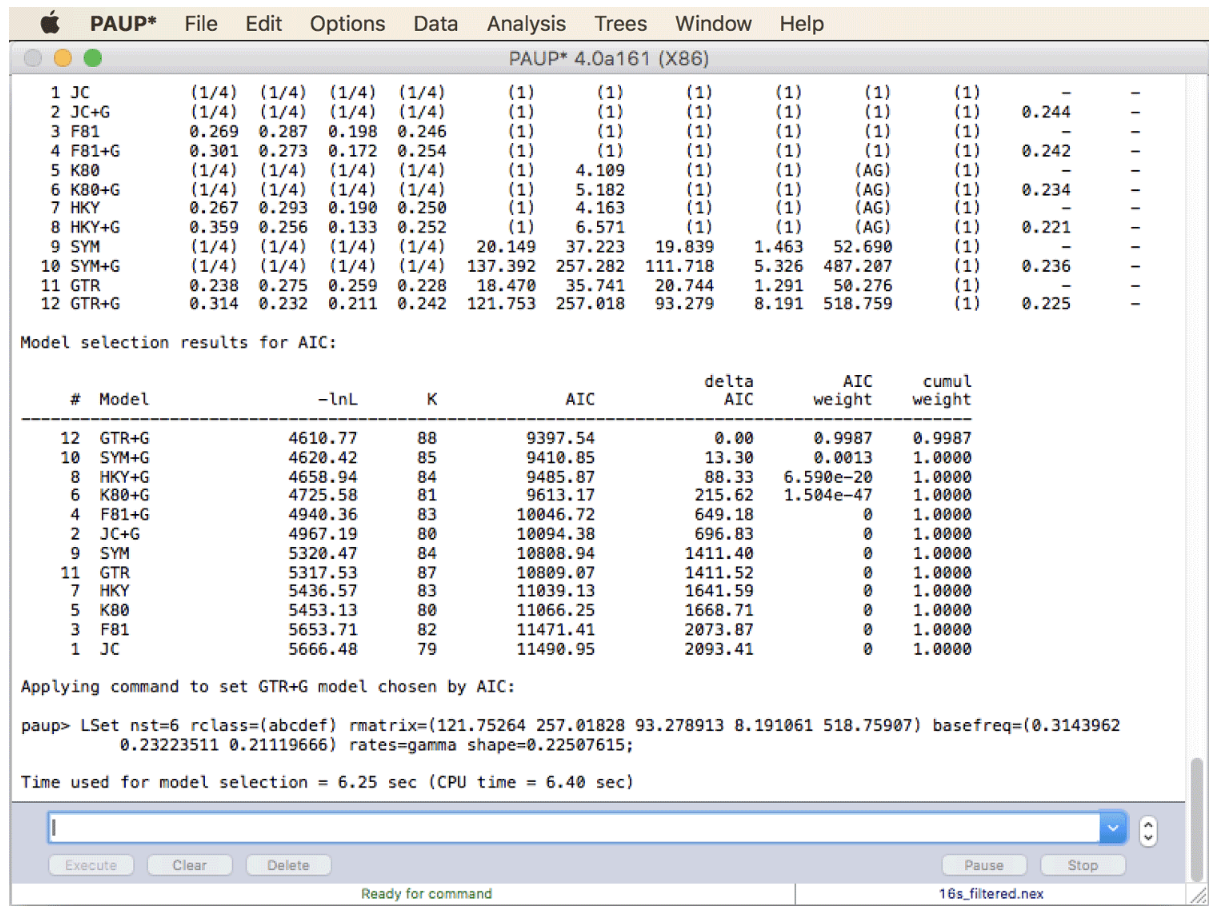
-
重复替换模型与 RAG1 序列比对 (rag1_filtered.nex) 的比较。
动动您发财的小手点个赞吧!
Reference
Source: https://github.com/mmatschiner/tutorials/blob/master/substitution_model_selection/README.md
本文由 mdnice 多平台发布