将群体的遗传图谱构建完成之后,我们得到的是一个excel文件,一堆数据,那么在做工作汇报或者发表文章的时候,我们必须要将数据进行可视化,最传统的方法当然是使用mapchart,我一直也是使用mapchart,其将遗传图谱可视化的效果图如下:

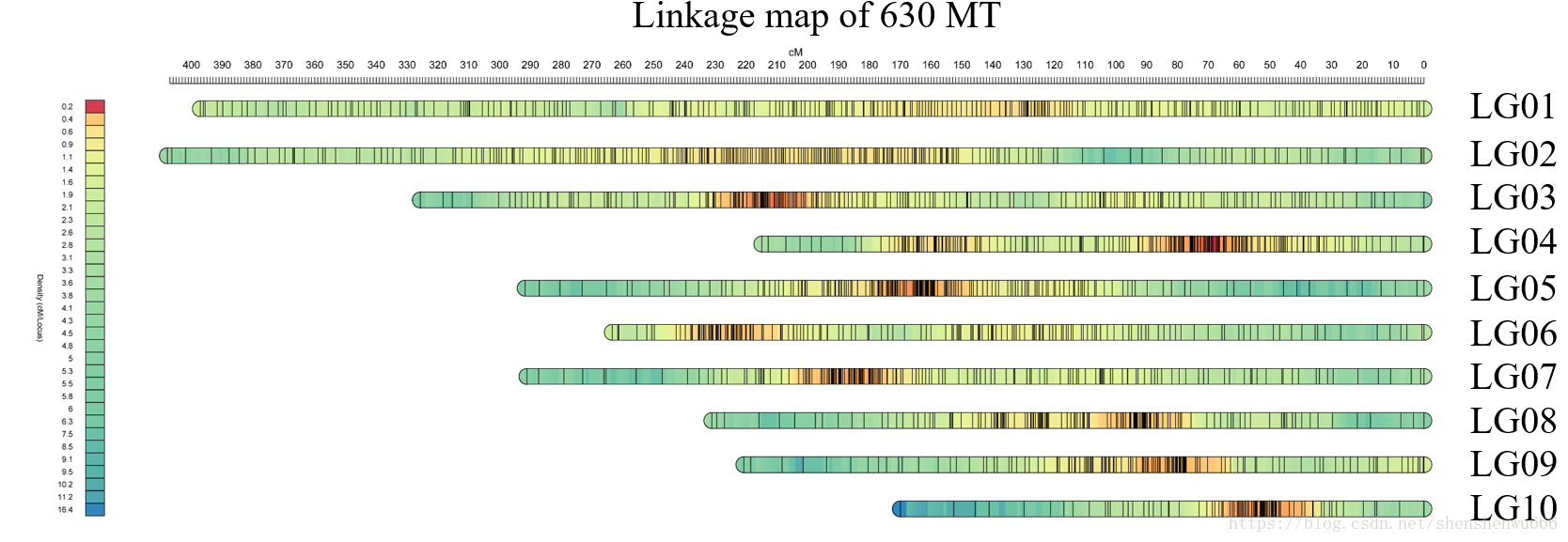
作为一个极度老牌的可视化软件,mapchart的弊端现在已经很明显了。在十几年前,一个群体的分子标记还很少,往往很普遍的情况是只有几百个SSR标记,而现在,随着测序行业的发展,随着SNP标记的普及,分子标记也越来越多,当分子标记多起来之后,mapchart的可视化结果就会很难看,很拥挤,很密集,甚至我用自己的2378个标记可视化时,出现了页面篇幅容纳不下这么多标记的情况,在无法使用mapchart可视化遗传图谱的情况下,我寻找到了另一种可视化遗传图谱的方法,它既能展示出每个标记在遗传图谱上的位置,同时还能用热图的形式展示出标记在遗传图谱上的密度。
我使用的是一个叫“LinkageMapView”的R包,此包既能在windows下使用,也同时支持linux和Mac。需要的输入文件格式如下:
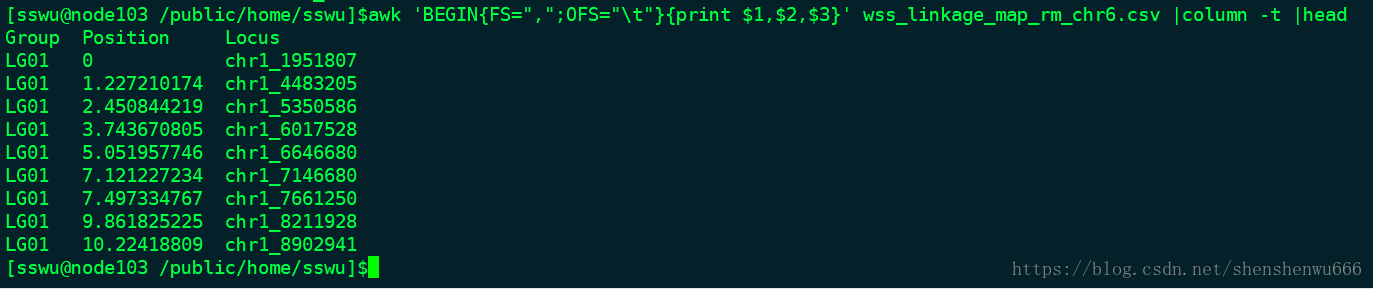
文件分三列,第一行是header,三列分别为Group、Position、Locus。group代表的是遗传图谱上的连锁群,position指的是遗传图谱上每个标记的遗传距离,locus指的是标记的名字。
以我的数据为例,我的输入数据是玉米的遗传图谱,共有10个连锁群,代码如下:
library(LinkageMapView)
a <- read.csv("C:\\Users\\Shenshen Wu\\Desktop\\linkage.csv")
maxpos <- floor(max(a$Position[a$Group == "LG01" | a$Group == "LG02" | a$Group == "LG03" | a$Group == "LG04" | a$Group == "LG05" | a$Group == "LG06" | a$Group == "LG07" | a$Group == "LG08" | a$Group == "LG09" | a$Group == "LG10"]))
at.axis <- seq(0, maxpos)
axlab <- vector()
for (lab in 0:maxpos) {
if (!lab %% 10) {
axlab <- c(axlab, lab)
}
else {
axlab <- c(axlab, NA)
}
}outfile = file.path("C:\\Users\\Shenshen Wu\\Desktop", "LinkageMap.pdf")
lmv.linkage.plot(a,outfile,mapthese=c("LG01","LG02","LG03","LG04","LG05","LG06","LG07","LG08","LG09","LG10"),denmap=TRUE, cex.axis = 1, at.axis = at.axis, labels.axis = axlab)
最后的可视化结果如下: