1. CDK (Chemistry Development Kit)
官网:https://cdk.github.io/点击打开链接
CDK是结构化学信息学和生物信息学的开源Java库。 该项目由Christoph Steinbeck,Egon Willighagen与Jmol和JChemPaint的开发人员Dan Gezelter于2000年发起。迄今为止,它是在科学界广泛支持下开展的最活跃的开源化学信息学项目之一。
CDK工具包为开发化学信息领域的新软件提供了许多功能,例如各种化学输入/输出(I/O)文件格式,包括简化的分子输入行输入系统(SMILES),化学标记语言(CML)和麻省理工学院设计语言(MDL); 结构发生器;;2D图表编辑和生成;3D几何图形生成;使用精确结构和SMARTS类查询进行子结构搜索;定量构效关系分子描述子计算(QSAR)研究;指纹计算;国际化学标识符(InChI)支持;在生物信息学领域,功能包括同源配体检测,代谢物鉴定等。

2. RDKit
官网:http://www.rdkit.org/点击打开链接
RDKit在2000-2006年期间在Rational Discovery开发和使用,用于构建吸收、分布、代谢、代谢、毒性和生物活性的预测模型。2006年6月Rational Discovery被关闭,但该工具包在BSD许可证下作为开源发布。目前,RDKit的开源开发由诺华积极贡献,其中包括诺华捐赠的源代码。
RDKit提供各种功能,如不同的化学I/O格式,包括SMILES/SMARTS,结构数据格式(SDF),Thor数据树(TDT),Sybyl线符号(SLN),Corina mol2和蛋白质数据库(PDB)。子结构搜索; 标准SMILES; 手性支持;化学转化;化学反应;分子序列化;相似性/多样性选择;二维药效团;分层子图/片段分析; Bemis和Murcko骨架;逆合成组合分析程序(RECAP); 多分子最大共同亚结构;功能图;基于形状的相似性;基于RMSD的分子分子比对;基于形状的对齐;使用Open3-DALIGN算法的无监督分子-分子比对;与PyMOL进行3D可视化集成;功能组过滤;分子描述符库;相似图;机器学习等等

3. Open Babel
官网:http://openbabel.org/wiki/Main_Page点击打开链接
Open Babel是一款开源自由软件,使用Open Babel可以将一种化学结构类型的文件格式转换成另一种文件格式,非常方便的进行各种类型的化学结构文件进行相互转换。
Open Babel由CC++编写,并提供C ++, Perl, Python等的API接口方便开发。
Open Babel 可以执行超过111种化学文件格式的读取、写入和相互转换,分别包括读取和写入82和85种文件格式。这些包括用于化学信息学(SMILES、InChI、MOL、MOL2),来自各种计算化学包(GAMESS、高斯、MOPAC)的I/O文件,晶体文件格式(CIF、ShelX),反应格式(MDL RXN ),分子动力学模拟(Amber)和对接软件包(AutoDock)使用的文件格式,2D绘图软件包(ChemDraw),3D查看器(Chem3D,Molden),化学动力学和热力学(ChemKin,Thermo)使用的格式。它支持使用Daylight SMARTS模式匹配过滤和搜索分子文件,并计算组贡献描述符,如LogP、极性表面积(PSA)和摩尔折射率(MR)。 它还提供可扩展的分子指纹和分子力学功能。

4. Cinfony
官网:http://cinfony.github.io/index.html#点击打开链接
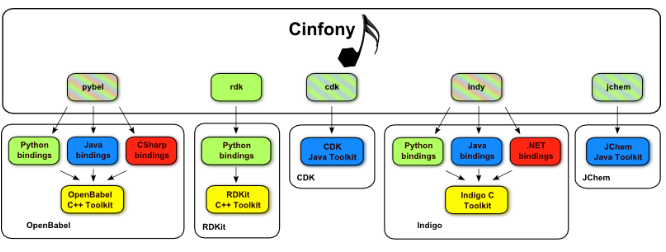
Cinfony是一个Python模块,它通过一种简单而强大的方法为Open Babel、RDKit和CDK提供了一个通用接口,以在这些工具包之间传递化学模型。它是Pybel的扩展,Pybel是一个只提供Open Babel访问权限的Python模块。它允许在应用程序编程接口(API)级别的互操作性,其优点是不需要对现有软件进行任何更改。它独立于平台,因此受到所有操作系统(如Mac,Windows和Linux操作系统)的支持,并且在BSD许可下作为开源发布。
5.Indigo
官网:http://lifescience.opensource.epam.com/点击打开链接
2009年,GGA软件服务根据GNU GPL条款发布了一个名为“Indigo”和相关软件的工具包。 它包含一些由GGA开发的独特算法,以及一些标准的着名算法。
Indigo是一个基于C ++语言的库,主要关注性能和基本化学特性。 围绕Python,Java和C#语言构建高级包装器或绑定。 这个库也允许多线程使用。 所有的二进制文件都受到所有基本操作系统的支持,即Windows,Linux和Mac OS X(32位和64位)。
支持常用和流行的化学形式:molfiles
- Rxnfiles v2000和v3000,SDF,RDF,SMILES,
- SMARTS和SMIRKS
- 支持四面体和顺式 - 反式立体化学
- 分子和反应呈现为PNG,SVG和PDF文件
- 分子和反应描述
- 芳香化和kekulization
- 规范(异构)SMILES计算
- 分子和反应的精确和亚结构匹配
- 支持匹配和突出显示
- 互变异构体和共振结构的匹配
- 计算分子和反应指纹
- 相似性搜索
- 最大公共子结构(MCS)算法
- R基团解卷积骨架检测
6.ChemoPy
官网:http://code.google.com/p/pychem/downloads/list点击打开链接
ChemoPy是用于计算常用结构和物理化学特征的开源软件包。它依赖于Pybel、RDKit、Open Babel和MOPAC等其他几个软件包,以提供其完整的功能。它计算出大约16个由19个各种特征组成的特征组,这些特征总共包含大约1,135个描述符。此外,它还提供了七种类型的分子指纹系统,包括拓扑指纹、电拓扑状态指纹、MACCS键、FP4键、原子对指纹、拓扑扭转指纹和Morgan /圆形指纹。 其中一些功能和指纹是使用Open Babel和RDKit派生的。使用MOPAC,ChemoPy计算大量的3D分子描述符。有趣的是,ChemoPy据报道是第一个基于MOPAC优化计算大量分子特征的开源软件包。
参考文献:Cao D-S, Xu Q-S, Hu Q-N, Liang Y-Z (2013) ChemoPy: freely available python package for computational biology and chemoinformatics. Bioinformatics 29:1092–1094.竟然来自母校开发。
7.ChemmineR
官网:http://www.bioconductor.org/packages/release/bioc/html/ChemmineR.html点击打开链接
ChemmineR是一种用于分析流行统计编程环境R中药物样小分子数据的cheminformatics包。该软件包的第一个版本于2008年出版。它包含用于化合物之间二维结构相似性比较的功能,针对化合物数据库的相似性搜索,用于聚类整个化合物库的功能,以及聚类结果的可视化。
2013年发布的最新ChemmineR版本具有其他实用程序和附加软件包,包括高效处理大量小分子的功能,物理化学/结构性质预测,结构相似性搜索,分类以及具有广谱的化合物文库的聚类包括用于成对比较的失配容忍MCS搜索算法。ChemmineR的当前版本还集成了Open Babel C ++库中实现的化学信息功能的一个子集。这些实用程序可以通过安装ChemmineOB软件包和Open Babel软件来启用。 ChemmineR可以自动检测ChemmineOB并使用其额外的实用程序。流功能允许使用sdfStream功能处理数百万个分子。最近的增加还包括快速和高效的指纹搜索,支持使用原子对或PubChem指纹,并通过新的SMIset对象类和SMILES导入/导出功能改进SMILES支持。
8.Chemkit
官网:http://www.chemkits.com/点击打开链接
Chemkit是由Kyle Lutz开发的开源库,支持分子建模,化学信息学和可视化功能。 Chemkit提供的主要功能包括I/O化学文件格式(pdb、cml、cjson、cube、fhz、fps、inchi、mol、mol2、sdf、smi等),访问Web资源,计算各种分子描述符,自动原子分型和拓扑构建以及基于OpenGL的可视化。
Chemkit库是用C++语言编写的,它使用Qt框架来处理图形。它是根据BSD许可证发布的。它是一个跨平台的库,受Windows、Mac和Linux操作系统的支持。
9.DeepChem
官网:https://deepchem.io/点击打开链接
DeepChem旨在提供高质量的开源工具链,将药物开发、材料科学、量子化学和生物学方面的深度学习应用普及化。

其他
- jCompoundMapper
- BALL: Biochemical Algorithms Library
- SMSD:Small Molecule Subgraph Detector
- Chemf
- Rcpi:Compound-Protein Interaction with R