欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/134884889
在蛋白质复合物中,通过链间距离,可以计算出在接触面的残基与接触面的面积,使用 BioPython 库 与 SASA (Solvent Accessible Surface Area,溶剂可及表面积) 库。SASA 计算主要以 水分子 是否通过为基础,水分子的半径是1.4 A,直径是 2.8 A,所以以 2.8 A 的距离,判断链间是否连接。
相关函数:
- 获取 蛋白质复合物 的链列表,即
get_chain_ids() - 获取 残基-原子 字典,注意原子名称与 PDB 的名称可能不同,即
get_atom_list() - 获取 全原子 列表,即
get_all_atoms() - 判断是否原子之间是否相连,即
is_contact() - 获取全部的接触面,原子计算残基维度,即
get_contacts() - 打印 ChimeraX 的显示命令,即
get_chimerax_cmd() - 端到端的计算接触残基,即
cal_chain_interface_residue() - 计算 SASA 面积,即
cal_sasa() - 获取复合物多链的子结构,即
get_sub_structure() - 端到端的计算接触面积,即
cal_chain_interface_area()
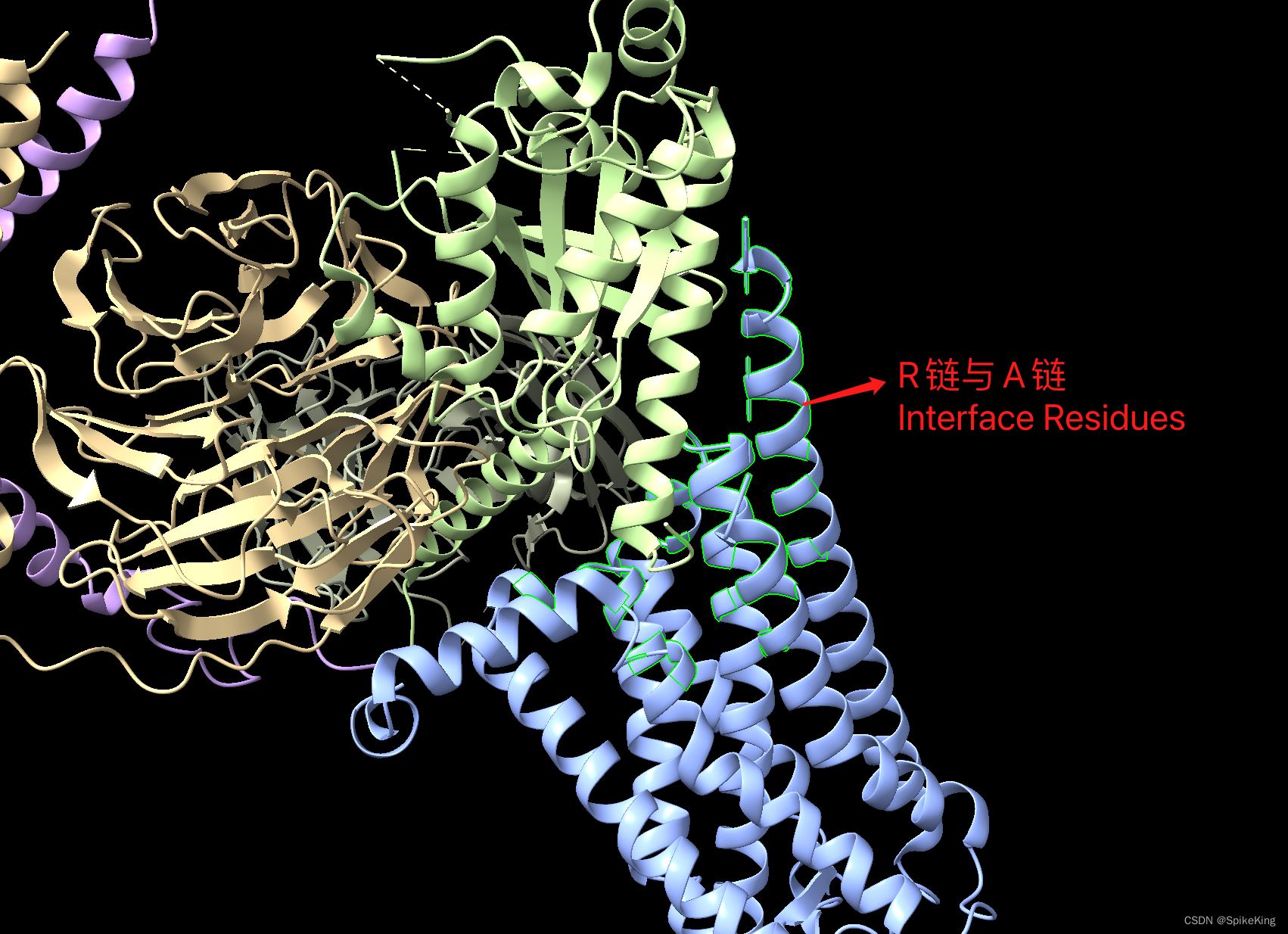
以 PDB 8j18 为例,计算链 A 与 链 R 的接触残基与面积:
源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2022. All rights reserved.
Created by C. L. Wang on 2023/12/8
"""
import os
from Bio.PDB import NeighborSearch
from Bio.PDB import PDBParser, Selection
from Bio.PDB.Chain import Chain
from Bio.PDB.Model import Model
from Bio.PDB.Structure import Structure
from root_dir import DATA_DIR
class MainComplexChainDist(object):
"""
复合物,多链距离计算
"""
def __init__(self):
pass
@staticmethod
def get_chain_ids(structure):
chain_ids = []
for chain in structure.get_chains():
chain_ids.append(chain.id)
return chain_ids
@staticmethod
def get_atom_list(structure, chains):
"""
获取 atom list 列表
注意: 输出的 atom 名字与 PDB 文件中的 atom 名字略有差异,例如
8jtl 第1个,PRO: CG 对应 OG1,CD 对应 CG2
8jtl 第2个,VAL: CG1 对应 CD1,CG2 对应 CD2
"""
output = dict()
for chain in structure:
if chain.id in chains:
for residue in chain.get_residues():
het_flag, res_seq, i_code = residue.get_id()
the_id = (chain.id + "_" + str(res_seq) + "_" + i_code).strip()
for atom in residue.get_unpacked_list():
if het_flag == ' ':
if the_id in output:
output[the_id].append(atom)
else:
output[the_id] = [atom]
return output
# Filter out all the atoms from the chain,residue map given by residue_map
@staticmethod
def get_all_atoms(residue_map):
all_atoms_out = []
for residue in residue_map:
for atom in residue_map[residue]:
all_atoms_out.append(atom)
# Set the b-factor to zero for coloring by contacts
atom.set_bfactor(0.0)
return all_atoms_out
@staticmethod
def is_contact(res_1, other_atoms, cutoff):
for atom in res_1:
ns = NeighborSearch(other_atoms)
center = atom.get_coord()
neighbors = ns.search(center, cutoff) # 5.0 for distance in angstrom
residue_list = Selection.unfold_entities(neighbors, 'R') # R for residues
if len(residue_list) > 0:
return True
return False
@classmethod
def get_contacts(cls, struc, all_atoms, verbose, cutoff):
progress = 0
contacts = []
for residue in struc:
progress += 1
# if len(verbose) > 0:
# print(verbose, progress, "out of", len(struc))
atom_list = struc[residue]
outcome = cls.is_contact(atom_list, all_atoms, cutoff)
if outcome:
contacts.append(residue)
return contacts
@staticmethod
def get_chimerax_cmd(contacts, chain_id, pdb_id="2"):
"""
计算命令
"""
tmp_list = []
for item in contacts:
req_id = int(item.split("_")[1])
tmp_list.append(req_id)
contacts_gap = []
prev, post = -1, -1
for i in tmp_list:
if prev == -1:
prev = post = i
continue
if i - post == 1:
post = i
else:
contacts_gap.append(f"#{
pdb_id}/{
chain_id}:{
prev}-{
post}")
prev = post = i
cmd = "select " + " ".join(contacts_gap)
return cmd
@classmethod
def cal_chain_interface_residue(cls, structure, chain1, chain2, cutoff=7.5):
"""
计算复合物结构的链内距离
"""
chain_ids = cls.get_chain_ids(structure)
# ['A', 'B', 'G', 'R', 'S']
print(f"[Info] chain_ids: {
chain_ids}")
assert chain1 in chain_ids and chain2 in chain_ids
# C 表示 chain,即将 structure 展开 chain 的形式
# 同时支持 A, R, C, M, S
structure_c = Selection.unfold_entities(structure, target_level="C")
atom_dict1 = cls.get_atom_list(structure_c, chains=[chain1])
atom_dict2 = cls.get_atom_list(structure_c, chains=[chain2])
print(f"[Info] res_num: {
len(atom_dict1.keys())}")
all_atoms_1 = cls.get_all_atoms(atom_dict1)
all_atoms_2 = cls.get_all_atoms(atom_dict2)
cutoff = cutoff
contacts_1 = cls.get_contacts(atom_dict1, all_atoms_2, "First molecule, residue ", cutoff)
contacts_2 = cls.get_contacts(atom_dict2, all_atoms_1, "Second molecule, residue ", cutoff)
print(f"[Info] contacts_1: {
len(contacts_1)}")
print(f"[Info] contacts_2: {
len(contacts_2)}")
return contacts_1, contacts_2
@staticmethod
def cal_sasa(structure):
"""
计算单体的表面积
"""
import freesasa
structure = freesasa.structureFromBioPDB(structure)
result = freesasa.calc(structure)
# classArea = freesasa.classifyResults(result, structure)
return result.totalArea()
@staticmethod
def get_sub_structure(structure, chain_list):
"""
获取 PDB 结构的子结构
"""
new_structure = Structure(0)
new_model = Model(0)
# 遍历模型中的所有链
for chain in structure[0]:
# 获取链的ID
tmp_chain_id = chain.get_id()
if tmp_chain_id not in chain_list:
continue
# 创建一个新的结构对象,只包含当前链
new_chain = Chain(tmp_chain_id)
new_chain.child_list = chain.child_list
new_model.add(new_chain)
new_structure.add(new_model)
return new_structure
@classmethod
def cal_chain_interface_area(cls, structure, chain1, chain2):
sub_all_structure = cls.get_sub_structure(structure, [chain1, chain2])
sub1_structure = cls.get_sub_structure(structure, [chain1])
sub2_structure = cls.get_sub_structure(structure, [chain2])
v1_area = cls.cal_sasa(sub_all_structure)
v2_area = cls.cal_sasa(sub1_structure) + cls.cal_sasa(sub2_structure)
inter_area = max(int((v2_area - v1_area) // 2), 0)
return inter_area
def process(self, input_path, chain1, chain2):
"""
核心逻辑
"""
print(f"[Info] input_path: {
input_path}")
print(f"[Info] chain1: {
chain1}, chain2: {
chain2}")
chain1 = chain1.upper()
chain2 = chain2.upper()
parser = PDBParser(QUIET=True)
structure = parser.get_structure("def", input_path)
contacts_1, contacts_2 = self.cal_chain_interface_residue(structure, chain1, chain2, cutoff=7.5)
cmd1 = self.get_chimerax_cmd(contacts_1, chain1)
cmd2 = self.get_chimerax_cmd(contacts_2, chain2)
print(f"[Info] chimerax: {
cmd1}")
print(f"[Info] chimerax: {
cmd2}")
inter_area = self.cal_chain_interface_area(structure, chain1, chain2)
print(f"[Info] inter_area: {
inter_area}")
def main():
ccd = MainComplexChainDist()
# input_path = os.path.join(DATA_DIR, "templates", "8p3l.pdb")
# ccd.process(input_path, "A", "D") # 2562
# ccd.process(input_path, "A", "J") # 490
input_path = os.path.join(DATA_DIR, "templates", "8j18.pdb")
# ccd.process(input_path, "R", "S") # 0
ccd.process(input_path, "A", "R") # 1114
if __name__ == '__main__':
main()