
论文标题:Predicting drug–protein interaction using quasi-visual question answering system
一、问题
目前DPI(DTI)分为两类:基于物理的方法(分子对接molecular docking:应用物理启发式的预定能量函数评估atom-level评估drug-protein interaction,但是难以评估构象熵和溶剂贡献,精度有限,对结构波动敏感,不能很好处理蛋白质的灵活性);基于机器学习的方法(将配体、蛋白质、蛋白质-配体相互作用统一到同一模型,回归/分类)
药物分子线性表示(SMILES)一般少于100个重原子,结构小。蛋白质通常包含1000+个重原子。而且一维蛋白质预测三维结构是一个难题,因此一维蛋白质结构无法捕捉空间信息。虽然有研究直接输入蛋白质3D结构,但是精度低(随着AlphaFold2的出现,这个问题应该有缓解)。同时高质量的蛋白质结构有限。
蛋白质可以由二维成对距离图替代表示(距离图可以用于生成和比较蛋白质的3D结构)。距离图是通过构成蛋白质的氨基酸之间的成对接触来紧凑地表示蛋白质的三维结构。
利用二维距离图来表示蛋白质,因此DPI任务可以视为视觉问答(VQA),但是有区别:
首先,在许多VQA场景中,图像大小可以调整为固定值,但是成对距离图的每个像素表示一对氨基酸之间的关系,如果对图进行下采样,则会导致信息丢失。
其次,SMILES的语法与自然语言不同,这使用自定义的tokenizer过程和合适的模型来获取分子线性符号的语义特征。
第三,训练集仍然比其他应用程序小得多,这需要仔细设计网络
二、模型
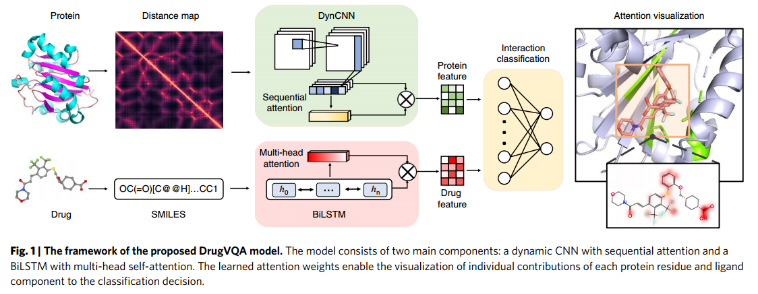
DrugVQA模型由两个主要部分:具有顺序注意的动态CNN(“Dynamic attentive CNN”)和具有多头自注意的BiLSTM(“Self-attentive BiLSTM”)。
1、Problem formulation
药物分子SMILES tokenizer:
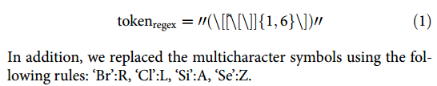

蛋白质可以简单地描述为一个由氨基酸残基列表P =(r1,…,rl)组成的线性序列,其中ri为i位氨基酸类型长度为20的one-hot向量,l为序列长度。2D pairwise distance map:

其中d(ri, rj)为残基i和残基j的Cα原子之间的距离,d0设为3.8Å,为相邻Cα原子之间的距离。因此距离矩阵为:
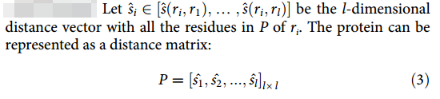
2、Dynamic attentive CNN
自适应CNN将蛋白质距离图编码为固定大小的向量表示。CNN模块residual blocks(from Resnet30,5×5卷积层+3×3卷积层,ELU代替ReLU) + sequential self-attention block。通常图像相同大小,但是蛋白质具有不同长度的氨基酸,并且无法缩放。

因动态神经网络,1)处理可变长度的输入,(2)预测每个氨基酸的重要性。首先删除残差块间的池化层,并对输入的两侧使用零填充,以确保残差块的结果与输入的大小相同。具体地说,给定一个蛋白质距离图P,最后一个残差块的输出仍然是l×l×N的维数,其中N是最后一个卷积层的通道数。然后使用平均池化对剩余块的信息输出进行压缩
3、Sequential attention
经过CNN特征提取得到的是:Pc,可以看作是蛋白质的序列表示,其中l是蛋白质中氨基酸(位点)的数量,Nf表示每个位点的空间特征:
![]()
由于大多数位点与药物结合没有直接关系,因此识别一小部分结合位点至关重要。为处理来自卷积层的不同大小的特征映射,并强调重要的结合位点,采用顺序自关注机制来充分利用这些特征进行分类。注意机制以Pc为输入,输出一个权重向量ap(蛋白质的注意矩阵),全连接层:

由于蛋白质结合口袋是由空间上相邻的多个连续位点组成的,进一步将wp2扩展为rp-by-dp矩阵,称为Wp2,以捕获结合口袋的整体结构信息,因此多头注意力:
![]()
式(5)可以看作是一个无偏置的双层MLP。通过将标注矩阵Ap与特征映射Pc相乘来计算rp加权和(注意力):
![]()
其中Pa是注意力特征图。Pa的大小为rp-by-nf,其中rp是一个可调的超参数,表示注意向量的数量。

4、Self-attentive BiLSTM
双向LSTM:

相互作用之间的关系:注意机制以整个LSTM隐藏状态H作为输入,输出一个权重向量Am(分子的注意矩阵)为(就是可学的线性注意力,而不是transformer中的注意力):

代码:
5、Classifier
Normalization(Pa+Ma)后 + 全连接层:


损失为交叉熵函数:
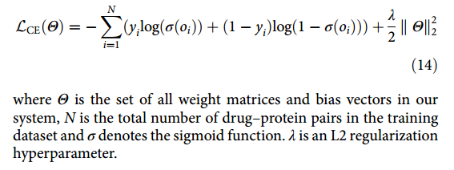
三、实验Experiments
1、Datasets
the directory of useful decoys, DUD-E, the human dataset and BindingDB
DUD-E:最终的数据集包含22,645个正例和1,407,145个负例。三折交叉验证。为了快速训练模型,使用一个平衡集(每个目标都是正的,随机选择等效的负的)进行训练,但使用整个集(不平衡的)进行评估。DUD-E: A Database of Useful (Docking) Decoys — Enhanced
Human:使用了一个平衡的数据集,其中阳性和阴性样本的比例为1:1。人类数据集包含5423种相互作用和1803种独特的蛋白质。使用80%/10%/10%训练/验证/测试随机分割。https://github.com/masashitsubaki/CPI_prediction/tree/master/dataset
BindingDB:包含来自BindingDB的39747个正例和31218个负例。训练集(50,155个交互),验证集(5607个交互)和测试集(5508个交互)。https://github.com/IBMInterpretableDTIP
2、Evaluation metrics
AUC-ROC。
对于Human,报告精度和召回值。
对于DUD-E,报告ROC富集度量(RE)。具体来说,RE评分被定义为在给定FPR阈值下的真阳性率与假阳性率(FPR)之比。在这里,报告了0.5%、1%、2%和5% FPR阈值下的RE得分。
3、消融实验

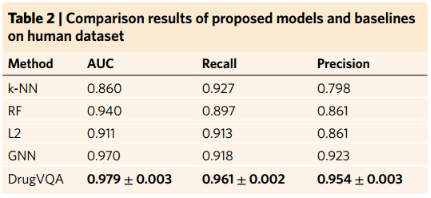
4、Comparisons on the human dataset
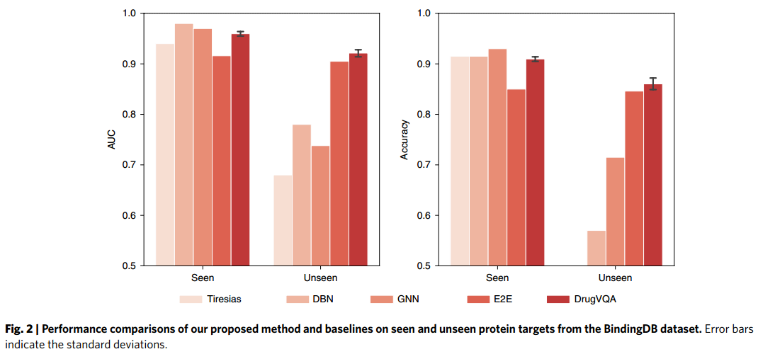
5、Comparisons on the BindingDB dataset
6、Comparisons on the DUD-E dataset.

7、Attention visualization.
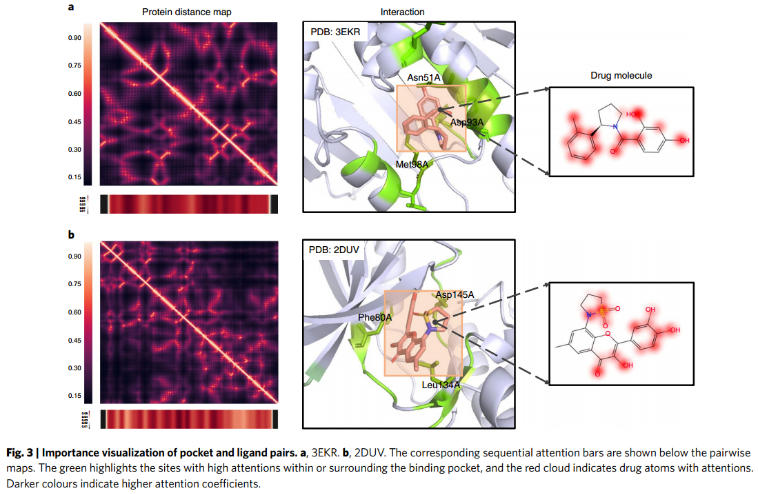
对于蛋白Hsp90(图3a),注意条突出显示了Asn51A、Asp93A和Met98A残基,它们与PDB: 3EKR中观察到的关键口袋残基高度重叠。对于蛋白CDK2(图3b),重要性图中突出显示的残基(Phe80A, Asp145A, Leu134A)和配体官能团与2DUV中观察到的相互作用高度相似。