Spateo工具使用指南(二)
Spateo工具使用指南(一)
近期发现Spateo这个神奇的工具能做到细胞分割,里面也有很多有趣的算法
RNA分割
在本教程中,我们将假设我们只有没有任何染色的 RNA 图像,并尝试使用 RNA 信号来识别单个细胞。
这是通过以下步骤完成的。
使用核定位基因识别细胞核(在我们的例子中,我们将使用Malat1和Neat1基因)。
使用未剪接的 RNA 识别额外的细胞核。
[可选] 将细胞核标签扩展到细胞质。
import spateo as st
import matplotlib.pyplot as plt
st.config.n_threads = 8
%config InlineBackend.print_figure_kwargs = {
'facecolor' : "w"}
%config InlineBackend.figure_format = 'retina'
加载数据
我们将使用Chen 等人于 2021 年截断的小鼠冠状切面数据集。
!wget "https://drive.google.com/uc?export=download&id=18sM-5LmxOgt-3kq4ljtq_EdWHjihvPUx" -nc -O SS200000135TL_D1_all_bin1.txt.gz
将下载的 UMI 计数和核染色图像加载到 AnnData 对象中。出于细胞分割的目的,我们将使用聚合计数矩阵,其中AnnData 的obs和var对应于空间 X 和 Y 坐标,矩阵的每个元素包含为每个 X 和 Y 捕获的 UMI 总数协调。
adata = st.io.read_bgi_agg(
'SS200000135TL_D1_all_bin1.txt.gz',
gene_agg={
'nuclear': ['Malat1', 'Neat1']} # Add a layer for nuclear-localized genes
)
adata
fig, axes = plt.subplots(ncols=3, figsize=(9, 3), tight_layout=True)
st.pl.imshow(adata, 'nuclear', ax=axes[0], vmax=2, save_show_or_return='return')
st.pl.imshow(adata, 'unspliced', ax=axes[1], vmax=5, save_show_or_return='return')
st.pl.imshow(adata, 'X', ax=axes[2], vmax=10)

用核定位基因识别细胞核
正如我们在上面观察到的,存在高和低 RNA 密度的区域。这需要将图像分成几个密度区域,然后分别对每个区域进行分割。否则,该算法可能会被错误校准,并且在 RNA 丰富的区域过于敏感,而在 RNA 稀疏区域过于严格。
我们建议首先从宽松开始(也就是将像素分成许多 RNA 密度区域),然后手动合并区域。
st.cs.segment_densities(adata, 'nuclear', 50, k=3, dk=3, distance_threshold=3, background=False)
st.pl.contours(adata, 'nuclear_bins', scale=0.15)
st.pl.imshow(adata, 'nuclear_bins', labels=True)
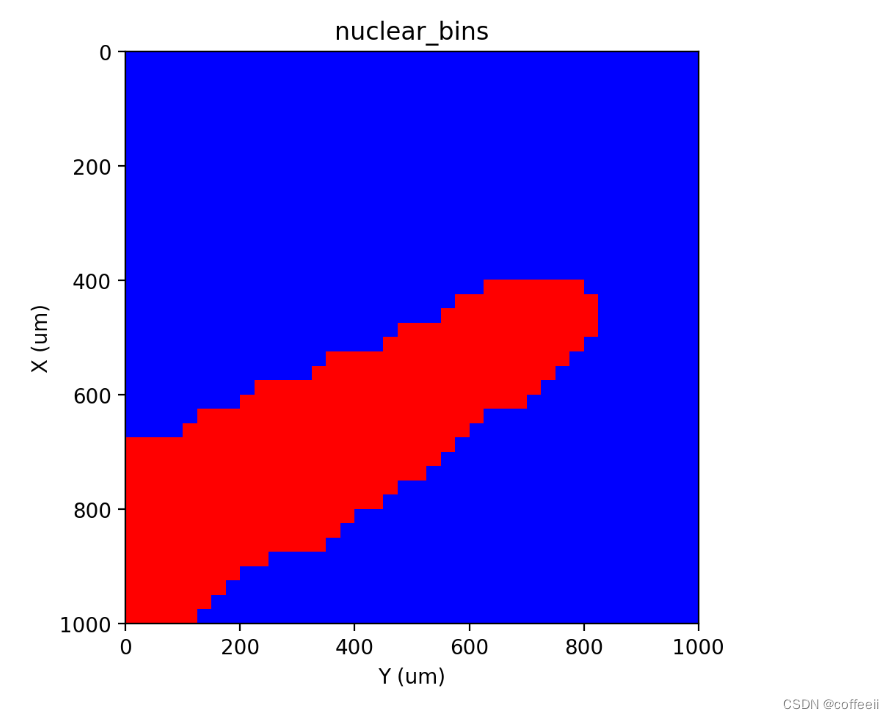
分割
st.cs.score_and_mask_pixels(
adata, 'nuclear', k=5, method='VI+BP',
vi_kwargs=dict(downsample=0.1, seed=0, zero_inflated=True)
)
st.pl.imshow(adata, 'nuclear_mask')
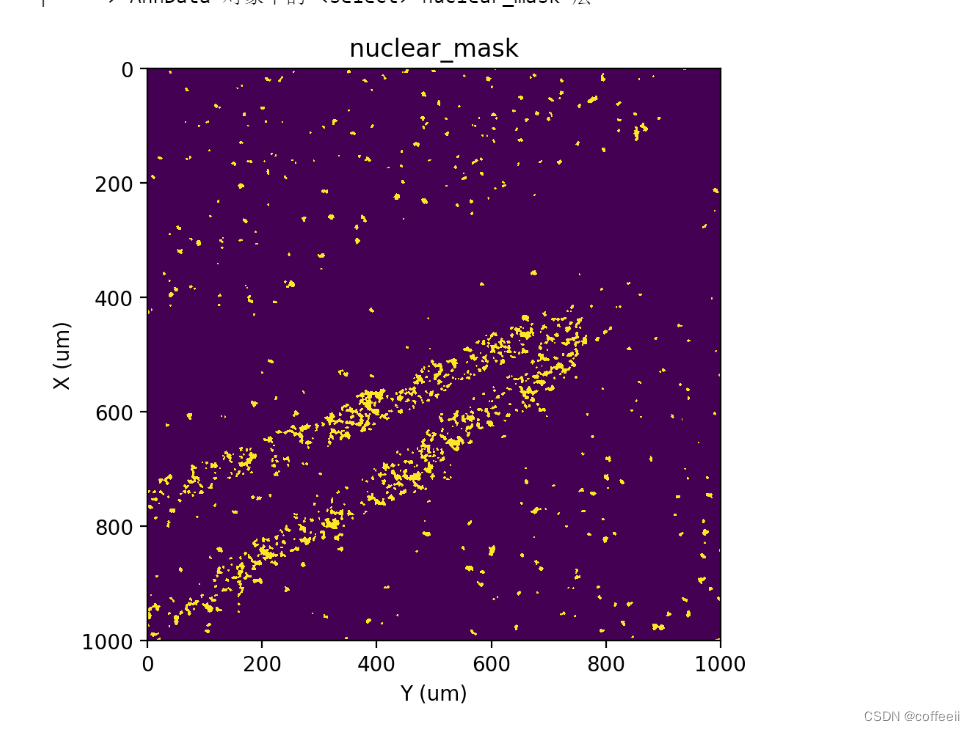
标签
st.cs.find_peaks_from_mask(adata, 'nuclear', 7)
st.cs.watershed(
adata, 'nuclear_distances', 1,
mask_layer='nuclear_mask',
markers_layer='nuclear_markers',
out_layer='nuclear_labels'
)
st.pl.imshow(adata, 'nuclear_labels', labels=True)
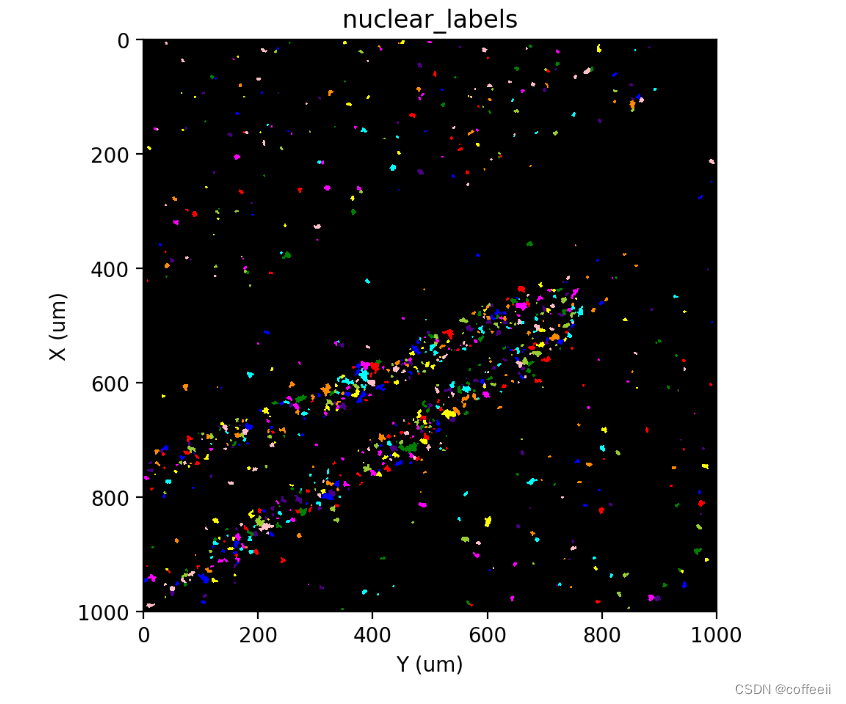
用未剪接的 RNA 识别额外的细胞核
st.cs.segment_densities(adata, 'unspliced', 50, k=3, dk=3, distance_threshold=3, background=False)
st.pl.contours(adata, 'unspliced_bins', scale=0.15)
st.pl.imshow(adata, 'unspliced_bins', labels=True)
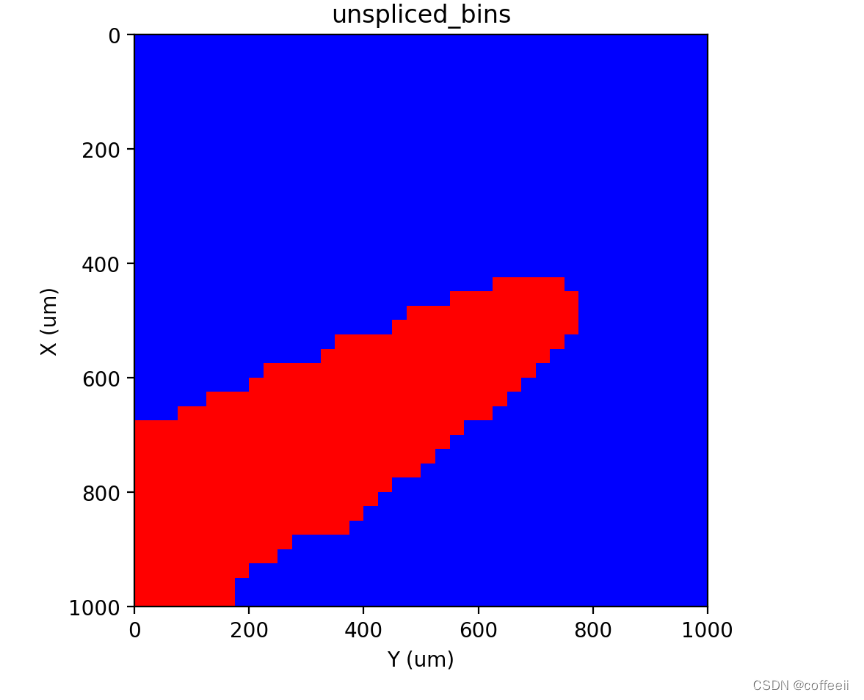
分割
然后,正如我们之前对核基因计数所做的那样,使用未剪接的 RNA 识别细胞核。请注意,该函数将自动检测 RNA 密度箱并相应地调整算法。另外,请注意,我们提供了certain_layer参数st.pp.segmentation.score_and_mask_pixels和seed_layer参数st.pp.segmentation.label_connected_components来指示从核定位基因获得的核标签。在内部,这些标签用于进一步帮助识别真实原子核。
st.cs.score_and_mask_pixels(
adata, 'unspliced', k=5, method='VI+BP',
vi_kwargs=dict(downsample=0.1, seed=0),
certain_layer='nuclear_labels'
)
st.pl.imshow(adata, 'unspliced_mask')
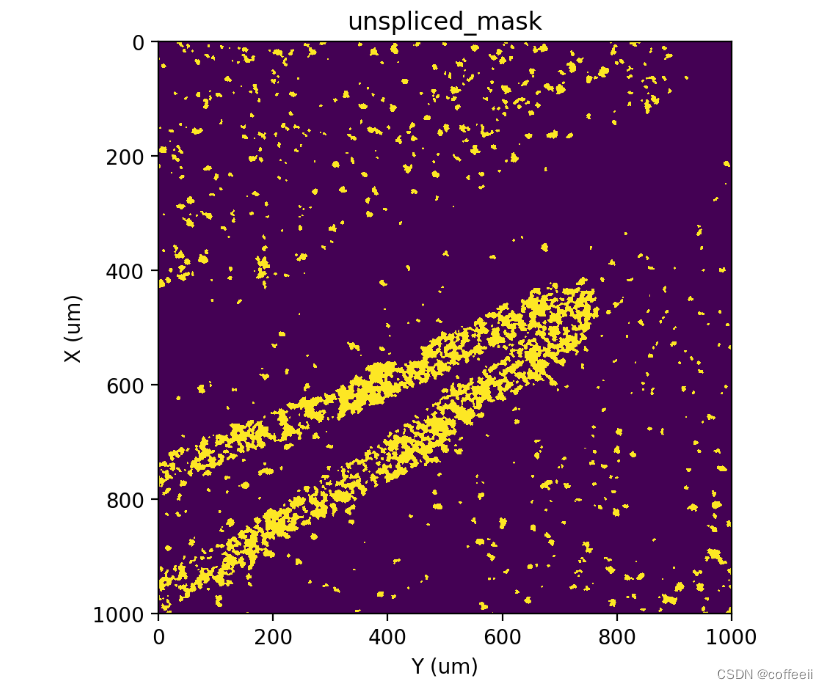
标签
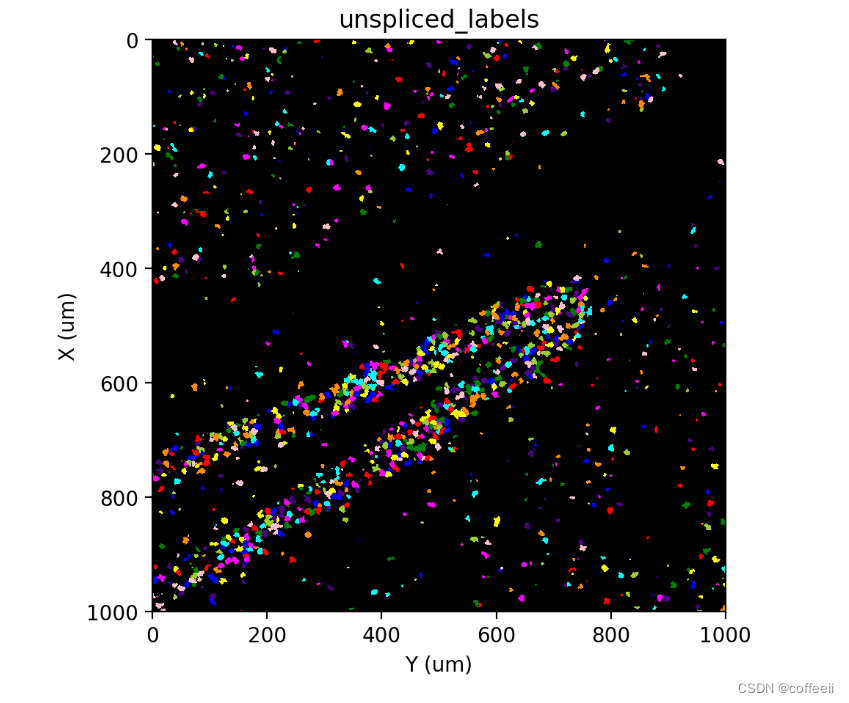
与之前我们使用核定位基因不同,这里我们知道一些我们想要保留的初始标签(在某些情况下,放大以填充上述掩码)。此外,使用未剪接的标签似乎会使一些 RNA 密集区域过度饱和,以至于很难分辨这些区域中细胞的边界。因此,我们不会使用尝试“填满”整个蒙版的分水岭方法,而是使用st.cs.label_connected_components限制每个标签可以分配的最大区域。
请注意,我们提供的seed_label参数是我们之前使用核定位基因获得的标签。
st.cs.label_connected_components(adata, 'unspliced', seed_layer='nuclear_labels')
st.pl.imshow(adata, 'unspliced_labels', labels=True)
更多生信知识欢迎交流v:coffeeiix(也可接单细胞转录组分析培训)