写在前面
昨天在绘制Circos圈图,已经隔了2年左右没有做这类的图了。这时间过得真是快,但是文章和成果依旧是没有很明显的成效。只能安慰自己,后面的时间继续加油吧!关于Cirocs图的制作,我从刚开始到现在都是是使用TBtools进行制作的,说实话,对于不想写代码的我(或是你)来说,生信可视化工具真的很方便。目前,生信可视化的工具很多,自己也加着2-3个类似的群,但最后一直使用到现在,以至于后面一直会使用的,TBtools是其中一个。我们在前面,也发表全程使用TBtools做分析教程,共线性分析 | Advance Circos图绘制、基于TBtools做基因家族分析。从使用者的角度来说,TBtools真心是一个不错的软件,基于开发者和用户的反馈和需求开发很多小软件。
关于今天的教程
我一直在说,我是一直在分享自己的学习笔记,所以内容等方面都是基于自己目前在学习内容,或是遇到的问题及解决方法。一方面是在记录自己的学习笔记,一方面是为了后续自己用到便于查找(我基本使用到需要的),最后是为了分享给需要的同学。但是,自己的能力有限,很多高深的内容自己涉及不到,或是没能力涉及。因此,也欢迎各老师或同学来投稿或分享你的学习笔记。
一个人的力量是有限的,但是一群人的力量是无法预测的!!
Cirocs教程分享
需要的文件
- 基因组长度文件
Chr1 56706830
Chr2 51972579
Chr3 58931556
Chr4 64763011
Chr5 44819618
Chr6 42866092
Chr7 56236587
Chr8 49719271
- 所需绘制文件的位置信息文件
Chr2 35739245 35739448 1 .
Chr2 36071610 36072481 1 -
Chr2 36199462 36199872 1 .
Chr2 36274372 36276705 1 -
Chr2 36443766 36444019 1 .
Chr2 39128193 39128397 1 .
Chr2 39485207 39485428 1 .
Chr2 41001395 41003552 1 +
基因组长度文件
- 打开TBtools中
Fasta stats

- 拖入基因文件和输出信息
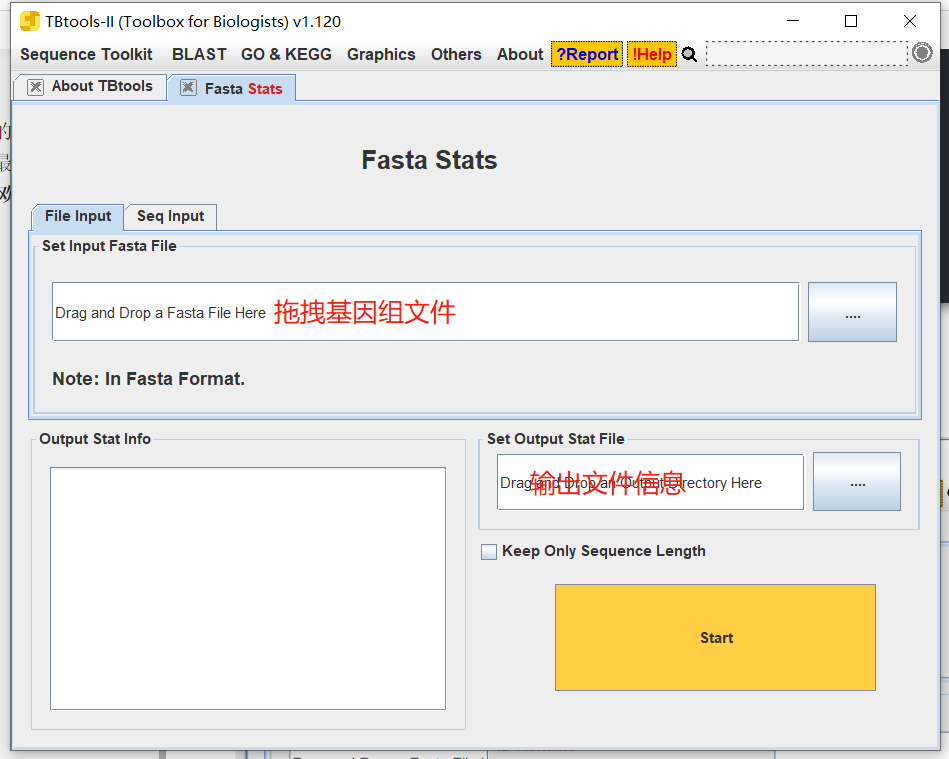
所绘制的基因的文件
- 方法1:直接提取,可以使用教程共线性分析 | Advance Circos图绘制的方法。
获得基因位置信息文件
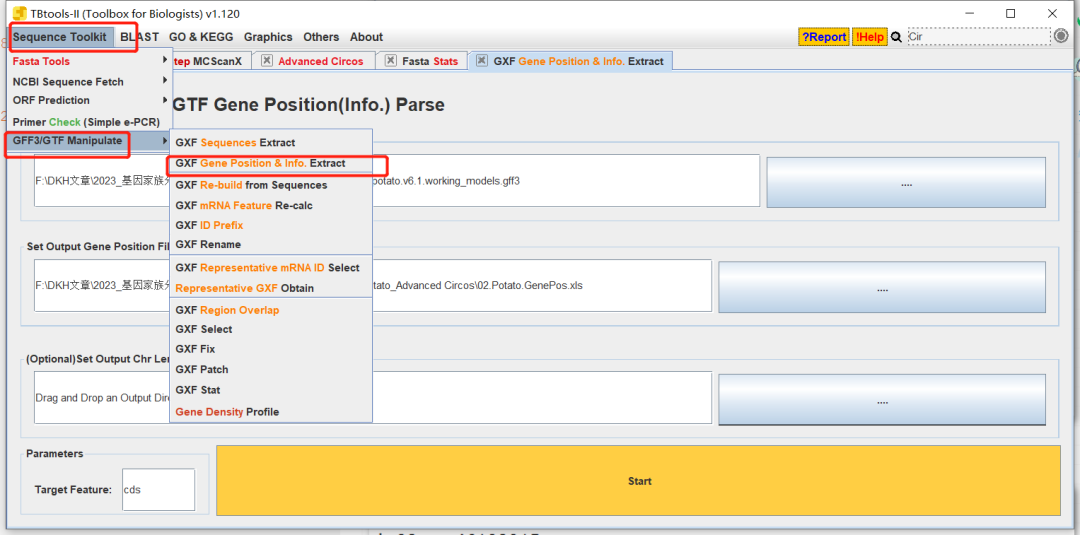
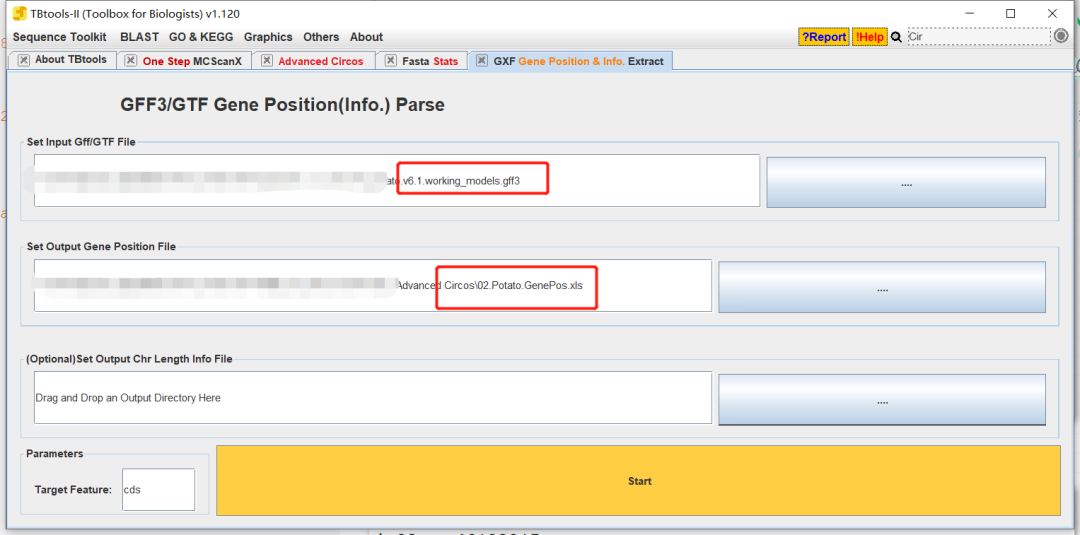
删除不必要的信息
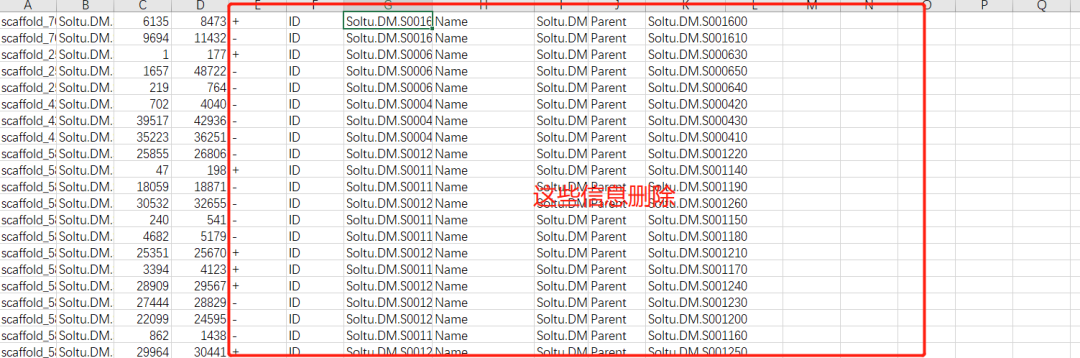
提取目标基因的信息


拖拽文件文件时,Selected Coluumn选择我们要match的列
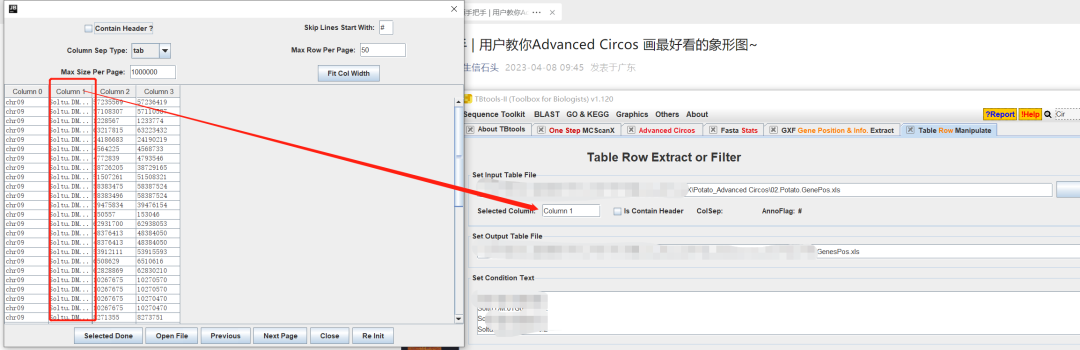
- 自己制作,所需的信息也就是的那么个,我们可以通过自己的注释文件进行提取就可以,使用
awk命令就医做简单的提取,我们这里就不在赘述。
我这里做个简单的记录,自己是补充前面做的分析的图,因此,自己手中并没有特定的绘图问题,只有一个总文件和所需绘图的基因ID,因此,只能用基因ID进行提取信息。
## 导入总文件
df <- read.table("all_lncRNA.bed.txt",header = T)
head(df)
##----
ID Chromosome Start End type Strand
MSTRG.247.1 Chr1 152110 154340 1 +
MSTRG.364.2 Chr1 1230854 1231704 1 +
MSTRG.410.1 Chr1 1536449 1536977 1 +
MSTRG.545.1 Chr1 2665821 2668057 1 +
MSTRG.545.2 Chr1 2665899 2667587 1 +
MSTRG.545.3 Chr1 2665902 2668057 1 +
(1)入所需基因ID文件,由于各列的长度不同,因此不能正常使用read.table或read.csv函数导入
df <- readLines("typeID.txt")
# 将每行数据按照空格或制表符进行拆分,得到一个列表
df.list <- strsplit(df,"\\s+")
# 计算最大列数,用于确定数据框的列数
max_cols <- max(sapply(df.list, length))
# 将数据补齐到相同的列数,用NA填充缺失值
df_matrix <- t(sapply(df.list, function(x) {c(x, rep(NA, max_cols - length(x)))}))
# 将数据补齐到相同的列数,用NA填充缺失值
data <- as.data.frame(df_matrix)
head(data)
colnames(data) <- c("C", "D", "H", "Ma", "O", "P", "S")
data <- data[-1,] ## 删除首行
head(data)
正常运行,应该有更简单的导入方法,欢迎交流。
(2)使用merge()函数,或是其他函数进行提取,其实merge在这里有点不太合适。
c <- as.data.frame(data$C)
c02 <- cold[!apply(is.na(c), 1, any),]
c03 <- as.data.frame(c02)
colnames(c03) <- c("ID")
head(c03)
df02 <- merge(df, c03, by = "ID")
head(df02)
write.table(df02, "C.bed.txt", sep = '\t',quote = FALSE, row.names = F, col.name = F)
此步仅记录自己本次做的过程,可忽略,亦可交流。
绘图
打开Advanced Circos
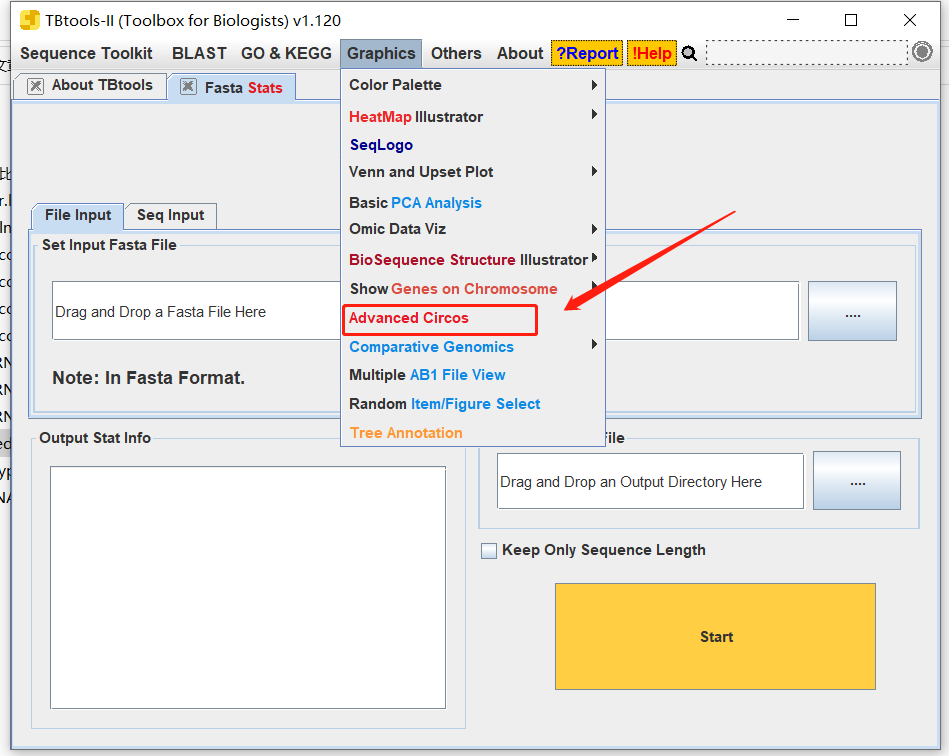
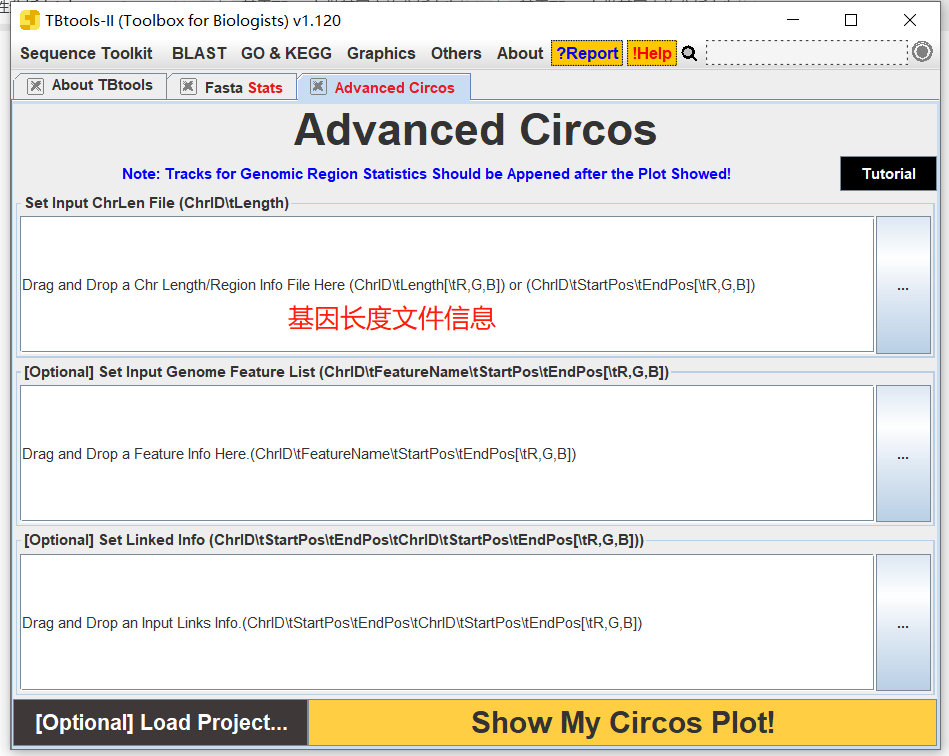
输入所需文件
这里,我就只需要输入基因长度文件信息即可
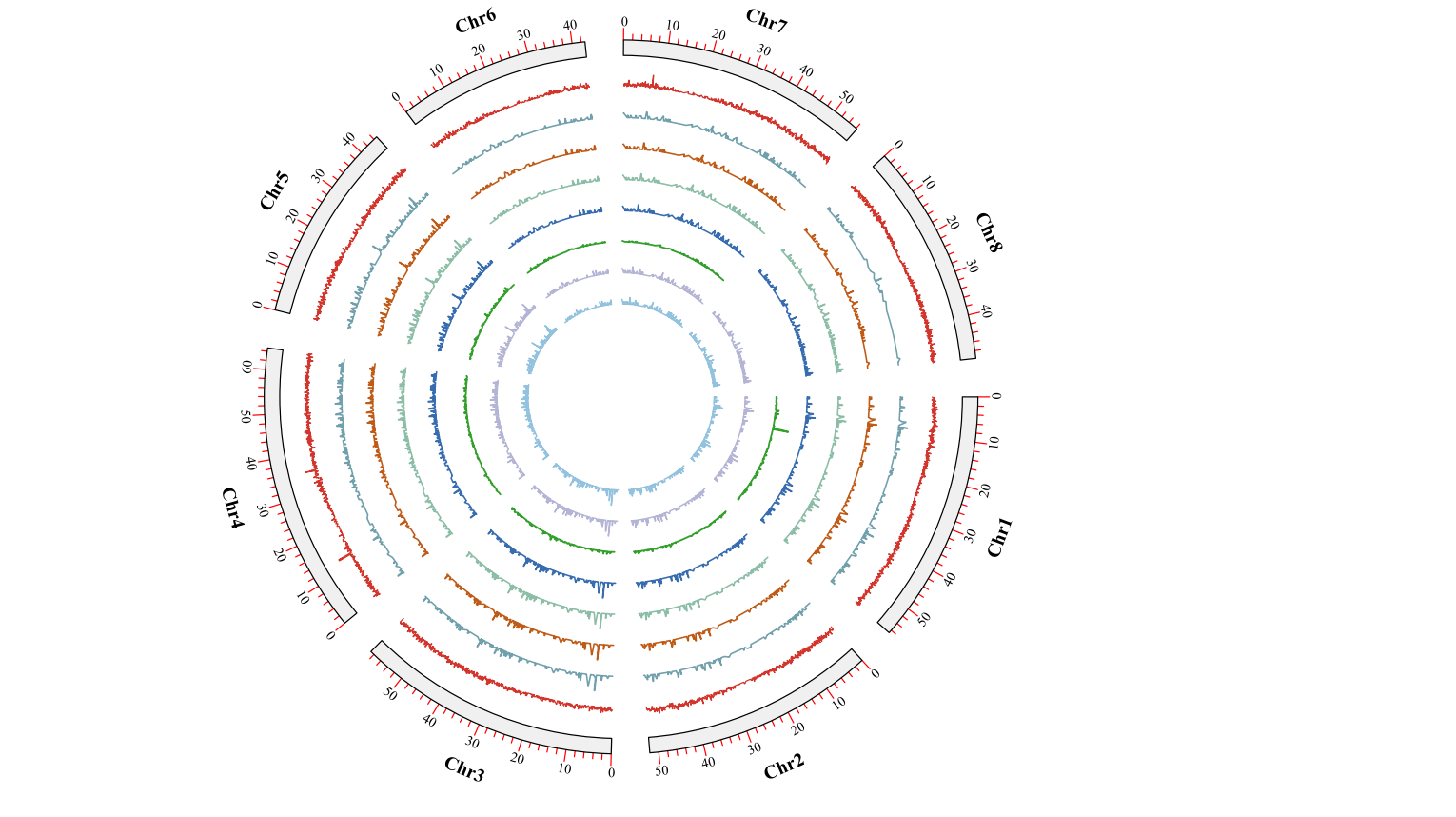
获得基因圈图
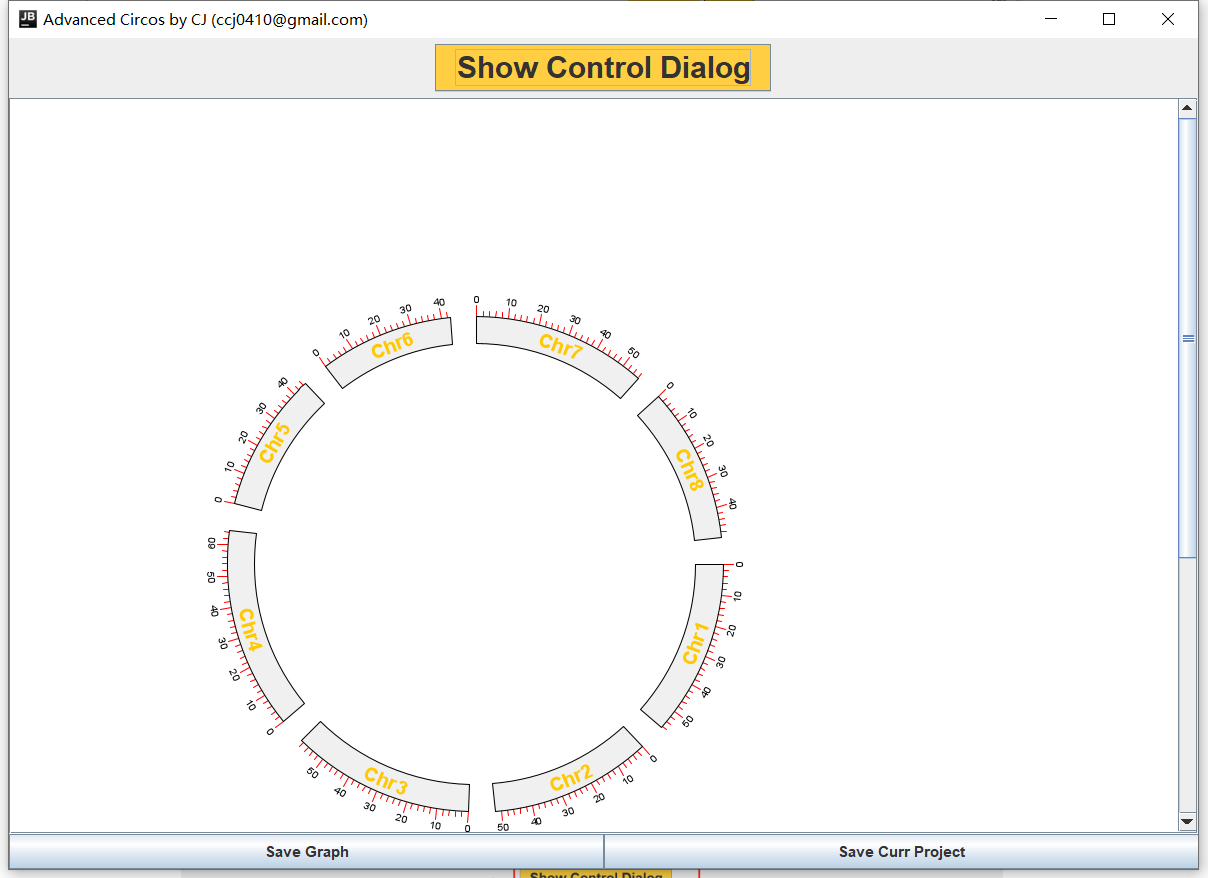
添加其他文件信息,点击Show Control Dialog

左边是调整基因圈图的参数,右边是添加其他信息和调整参数
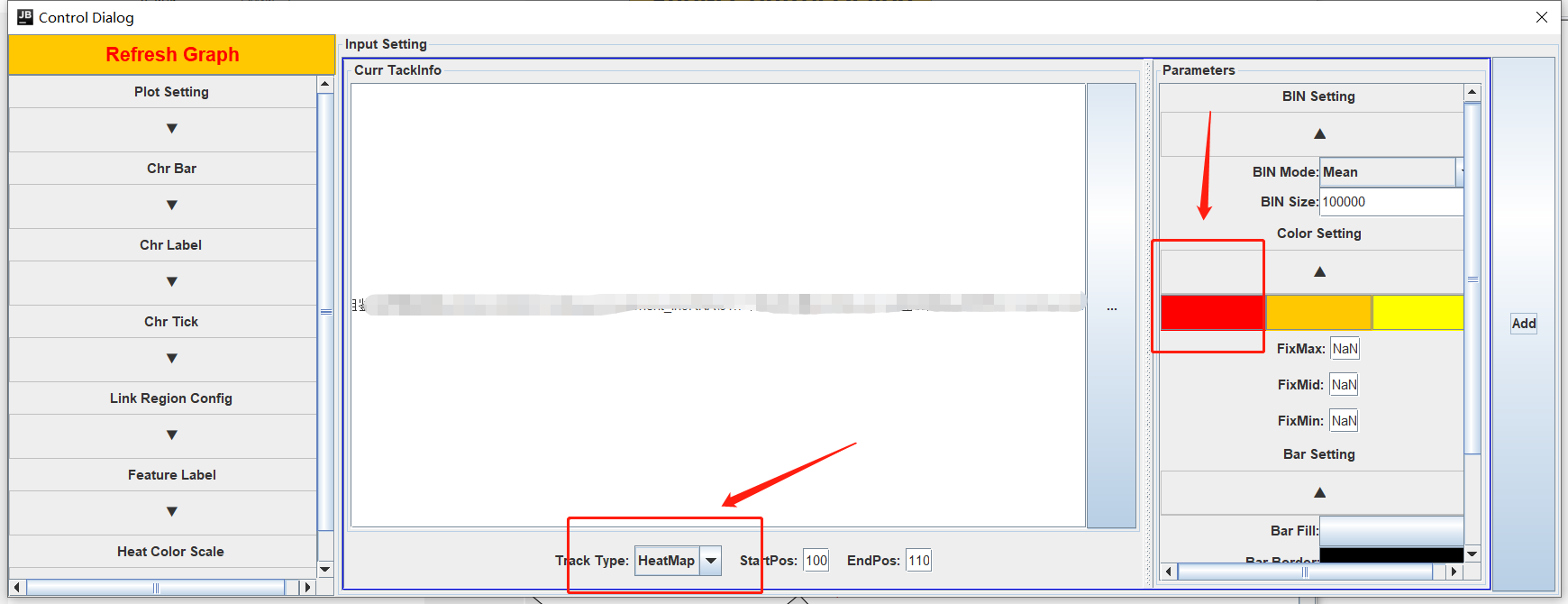
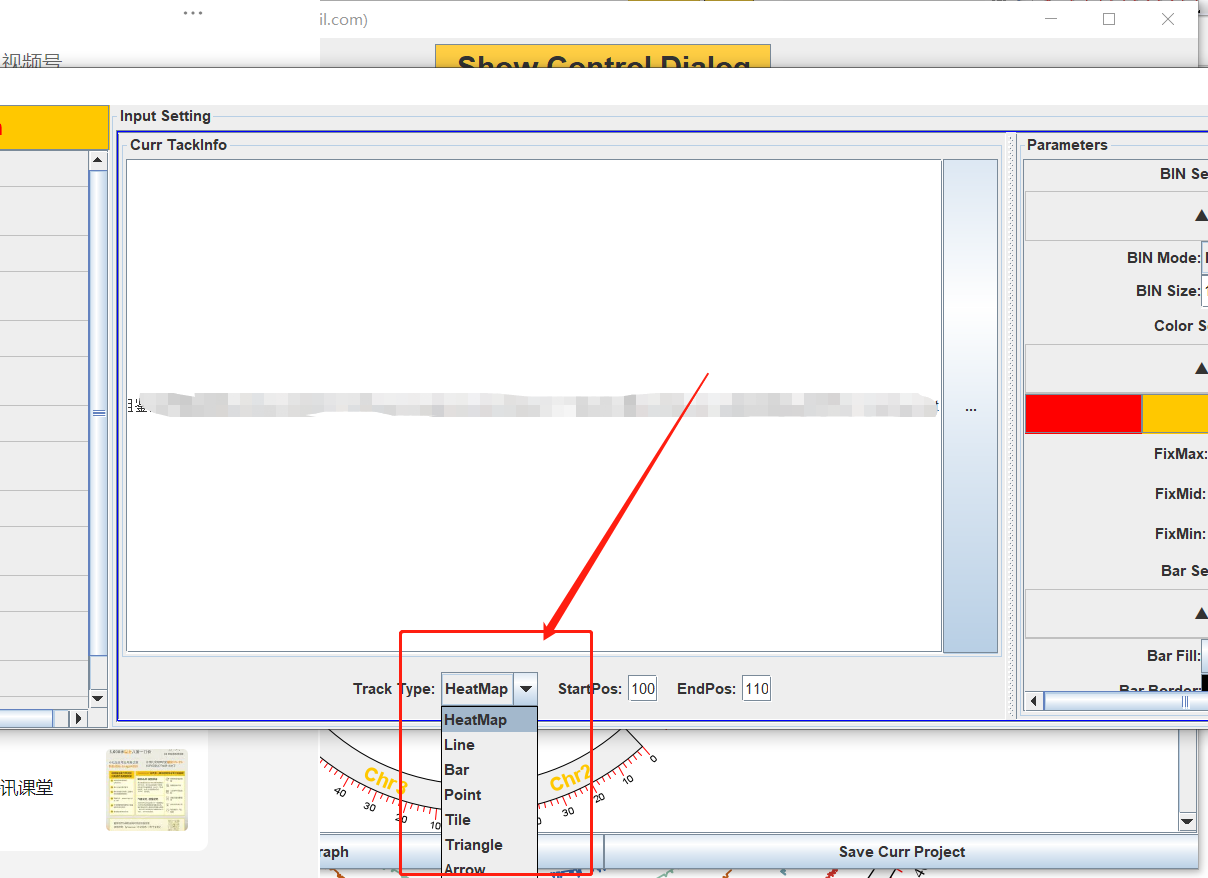
提供了很多图形的选项,根据自己的需求进行调整即可,以及颜色的调整。
最后就是细节调整,这些主要依赖于个人的审美和搭配。
---
**往期文章:**
**1. 最全WGCNA教程(替换数据即可出全部结果与图形)**
- [WGCNA分析 | 全流程分析代码 | 代码一](https://mp.weixin.qq.com/s/M0LAlE-61f2ZfpMiWN-iQg)
- [WGCNA分析 | 全流程分析代码 | 代码二](https://mp.weixin.qq.com/s/Ln9TP74nzWhtvt7obaMp1A)
- [WGCNA分析 | 全流程代码分享 | 代码三](https://mp.weixin.qq.com/s/rU76rLG4AayuiHbDhgOGBg)
----
**2. 精美图形绘制教程**
- [精美图形绘制教程](https://mp.weixin.qq.com/mp/appmsgalbum?__biz=MzAwODY5NDU0MA==&action=getalbum&album_id=2614156000866385923&scene=173&from_msgid=2455848496&from_itemidx=1&count=3&nolastread=1#wechat_redirect)
**3. 转录组分析教程**
小杜的生信筆記,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!