前言
近两天在做基因家族的分析,这是第一次接触这块,很多不是很了解。在筛选同源蛋白序列的时候,以前都是使用BLAST进行比对,这次看到很多教程都是使用HMMER进行寻找,那么也就顺便来学习一下吧。HMMER使用并不是很难,原理与Blast一致,其实对于初学来说,难的主要是建立
.hmm这一步。

Hmmer的安装
安装,主要是使用源码安装或是是使用conda进行安装即可。
conda安装
conda install -y hmmer
源码安装:
官网:http://www.hmmer.org/
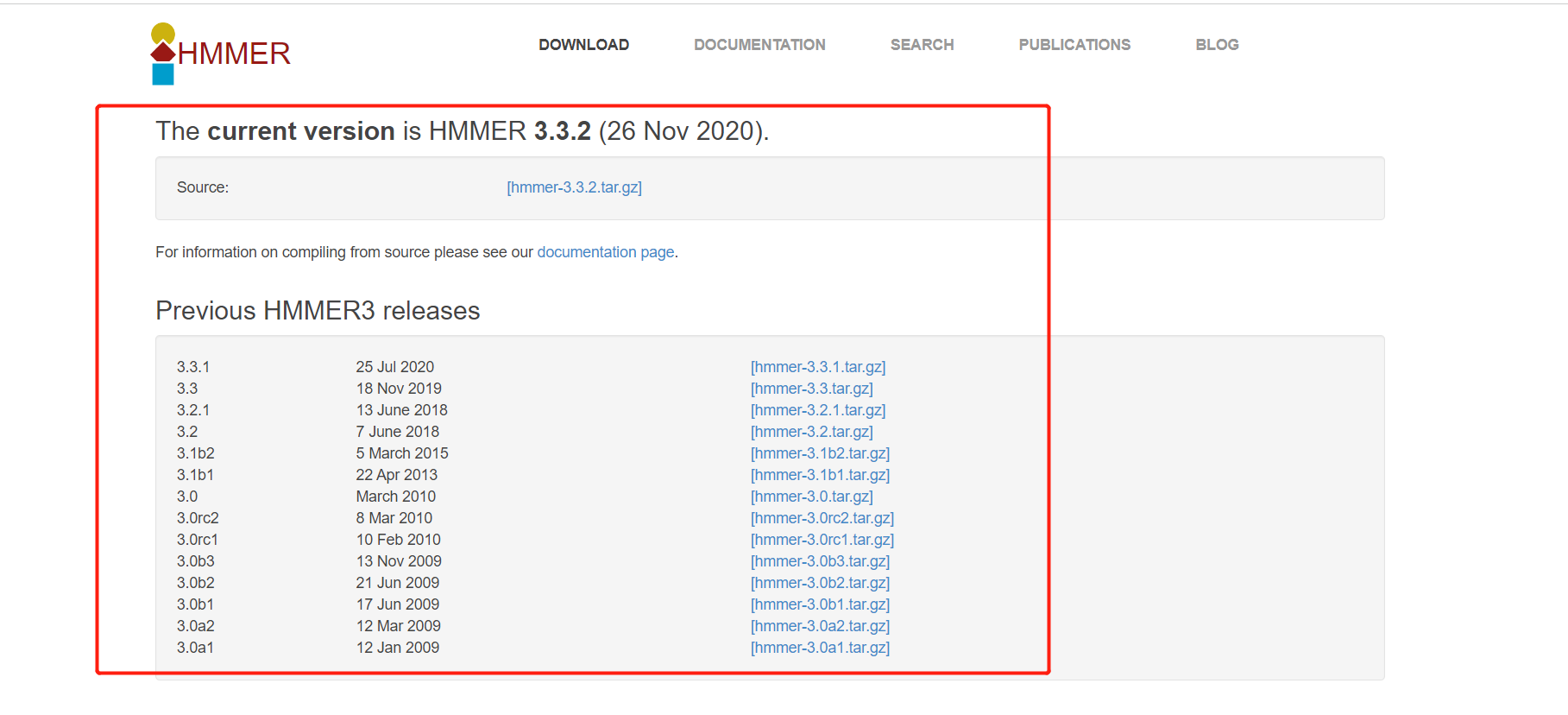
任意下载一个版本即可,安装步骤不再做说明。
使用hmmbuild构建.hmm文件
在有些数据库中是有.hmm文件,只需要下载即可。但是,这仅仅只限于有些大数据库。对于我们自己使用,不可能全部都有,这就需要我们自己构建,很多教程到这步就是让你收费了…。
在本教程,讲述其中一种方法吧,希望对大家有所帮助。
hmmbuild构建时,需要使用
.sto文件进行构建。因此,我们必须获得.sto文件。
1. 使用mafft对蛋白基因进行比对
- 安装(自己解决,类似的方法即可)
- 比对
mafft --auto --clustalout ../Pfam.fasta > Pfam.clustal
- 转换
我们需要将.clustal转换成.sto格式。我们推荐使用Fasta to Phylip Sequence Converter网站进行转换(http://sequenceconversion.bugaco.com/converter/biology/sequences/fasta_to_phylip.php)。
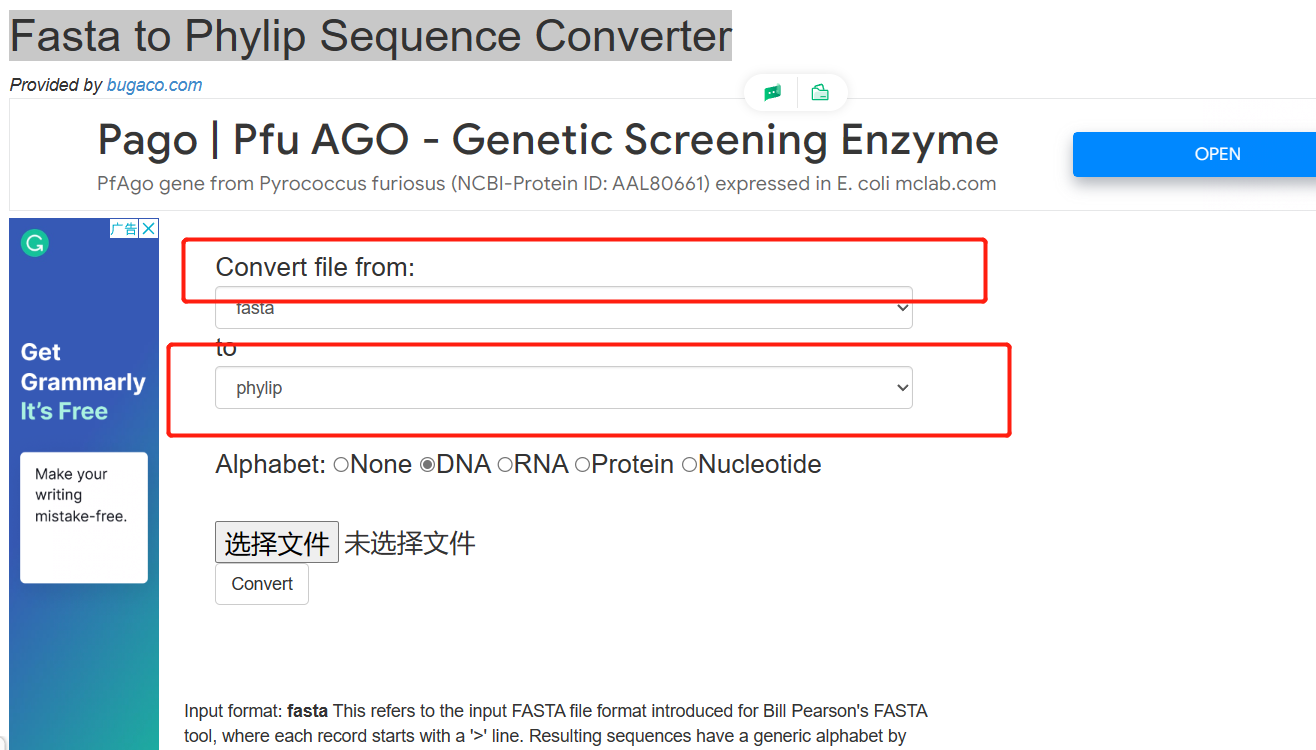

可以有这么多的格式进行相互转换。我们选择以下的格式进行转换即可。
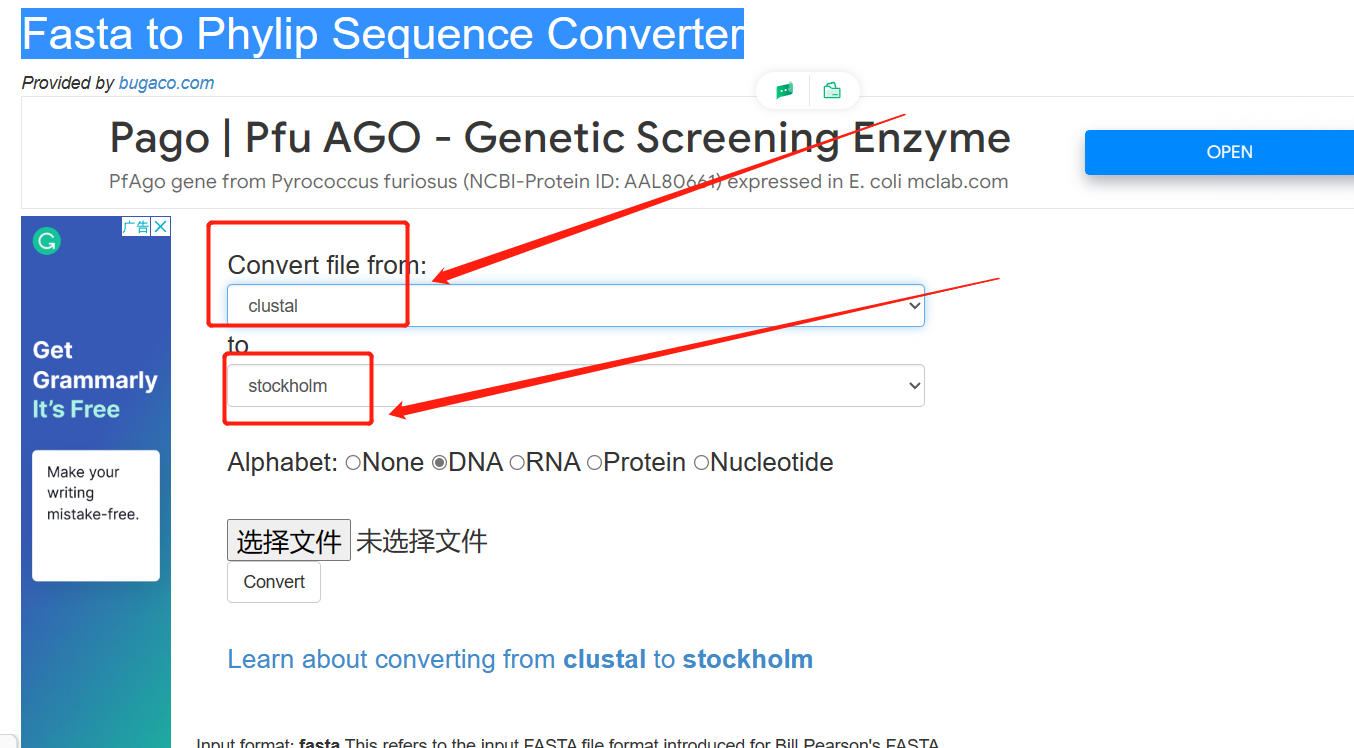
- clustat格式

- ** sto格式**
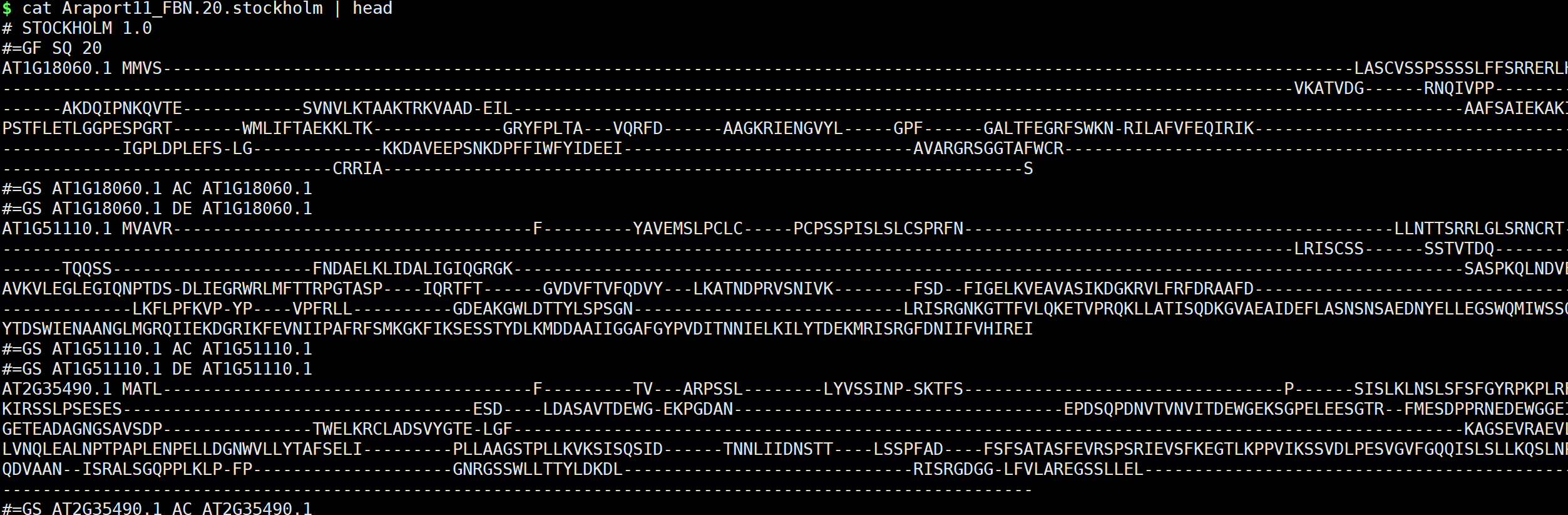
以上转换后就可以直接进构建.hmm文件
2. hmmbuild构建.hmm文件
hmmbuild Pfam.hmm Pfam.sto
构建运行还是很快的,主要还是取决于你的基因数量。

3. hmmersarch进行同源搜索
直接搜索即可,序列格式fa…等其他格式。我个人感觉其他格式,我们的用的还是很少吧,主要还是fa格式。
$ hmmsearch -h
# hmmsearch :: search profile(s) against a sequence database
# HMMER 3.3.2 (Nov 2020); http://hmmer.org/
# Copyright (C) 2020 Howard Hughes Medical Institute.
# Freely distributed under the BSD open source license.
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Usage: hmmsearch [options] <hmmfile> <seqdb>
Basic options:
-h : show brief help on version and usage
Options directing output:
-o <f> : direct output to file <f>, not stdout
-A <f> : save multiple alignment of all hits to file <f>
--tblout <f> : save parseable table of per-sequence hits to file <f>
--domtblout <f> : save parseable table of per-domain hits to file <f>
--pfamtblout <f> : save table of hits and domains to file, in Pfam format <f>
--acc : prefer accessions over names in output
--noali : don't output alignments, so output is smaller
--notextw : unlimit ASCII text output line width
--textw <n> : set max width of ASCII text output lines [120] (n>=120)
使用:
hmmsearch Pfam.hmm your.fa > restult.out.txt
结果文件直接输出系统默认的结果,主要是E-value值小于0.05,系统就默认保留。这部分可以根据自己的要求,进一步的过滤即可。
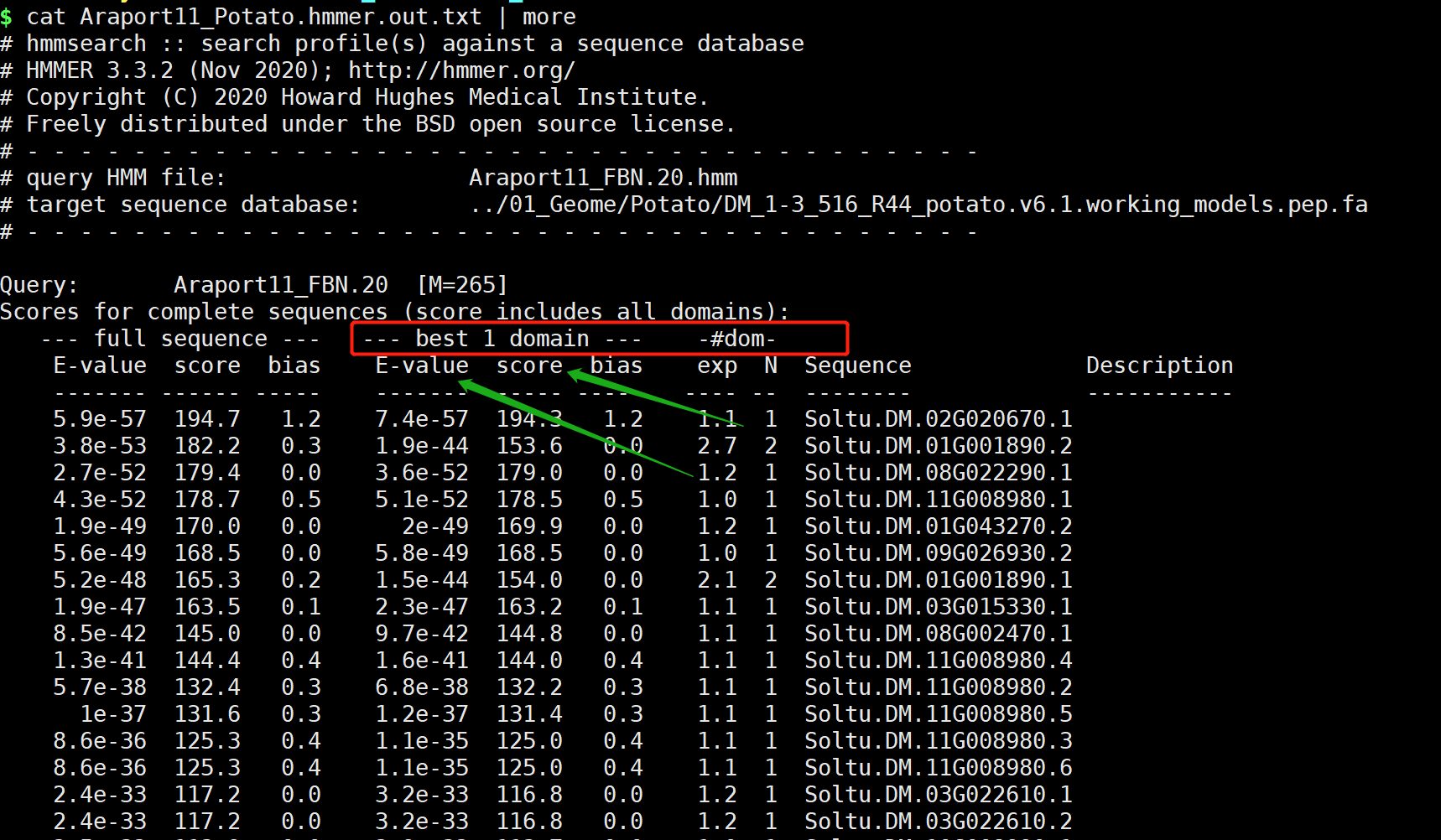
这基因就是我们Pfam数据库能同源搜索的基因。到这里你就是你需要的结果。
HMMER与BLAST之间的差异
-
- HMMER与BLAST之间的差异,HMMER只能搜索蛋白序列,BLAST范围更广。
-
- HMMER结果只有目标的同源基因(算是已经过滤一遍),BLAST结果是原始数据,需要后期进一步的过滤。BLAST结果会罗列出同源的全部基因之间的信息。
使用哪一个呢??
根据自己的需求即可。
教程简洁,希望对你有所帮助。
往期文章推荐:
-
- 转录组分析教程
小白学生信 | 转录组上游分析教程

- 转录组分析教程
-
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
- 2. 精美图形绘制教程
小杜的生信筆記,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!